Article Text

Abstract
Background Alveolar macrophages (AM) in COPD have fundamentally impaired responsiveness to Toll-like receptor 2 (TLR2) and TLR4 ligands of non-typeable Haemophilus influenzae (NTHI). However, the contribution of innate immune dysfunction to exacerbations of COPD is unexplored. We hypothesised that impaired innate AM responses in COPD extend beyond NTHI to other pathogens and are linked with COPD exacerbations and severity.
Methods AMs, obtained by bronchoalveolar lavage from 88 volunteers with stable-to-moderate COPD, were incubated with respiratory pathogens (NTHI, Moraxella catarrhalis (MC), Streptococcus pneumoniae (SP) and TLR ligands lipopolysaccharide, Pam3Cys) and elicited IL-8 and TNF-α were measured by microsphere flow cytometry. NF-κB nuclear translocation was measured by colorimetric assay. AM TLR2 and TLR4 expression was determined by immunolabeling and quantitation of mean fluorescent indices. Participants were monitored prospectively for occurrence of COPD exacerbations for 1 year following bronchoscopy. Non--parametric analyses were used to compare exacerbation-prone and non-exacerbation-prone individuals.
Results 29 subjects had at least one exacerbation in the follow-up period (exacerbation-prone) and 59 remained exacerbation-free (non-exacerbation-prone). AMs of exacerbation-prone COPD donors were more refractory to cytokine induction by NTHI (p=0.02), MC (p=0.045) and SP (p=0.046), and to TLR2 (p=0.07) and TLR4 (p=0.028) ligands, and had diminished NF-κB nuclear activation, compared with non-exacerbation-prone counterparts. AMs of exacerbation-prone subjects were more refractory to TLR2 upregulation by MC and SP (p=0.04 each).
Conclusions Our results support a paradigm of impaired innate responses of COPD AMs to respiratory pathogens, mediated by impaired TLR responses, underlying a propensity for exacerbations in COPD.
- COPD Exacerbations
- Cytokine Biology
- Innate Immunity
- Macrophage Biology
Statistics from Altmetric.com
Key messages
What is the key question?
-
Alveolar macrophage dysfunction and bacterial persistence promote progression of COPD. What is the contribution of innate immune dysfunction to the predilection for exacerbations of COPD?
What is the bottom line?
-
Alveolar macrophages of exacerbation-prone COPD donors were more refractory to cytokine induction by respiratory bacterial pathogens, through Toll-like receptor 2 (TLR2) and TLR4 signalling, and to microbial upregulation of TLR expression, than their non-exacerbation-prone counterparts.
Why read on?
-
This study supports a paradigm of impaired innate responses of COPD alveolar macrophages to respiratory pathogens, mediated by impaired TLR responses, underlying a propensity for COPD exacerbations.
Introduction
Exacerbations are major contributors to morbidity and mortality associated with COPD.1 Approximately half of the exacerbations are driven by acquisition of new strains of respiratory bacterial pathogens, and increased risk of COPD exacerbation is associated with increased mortality.2 ,3 The individual propensity for COPD exacerbations varies widely. However, the underlying biological basis for this predilection remains unknown.
We previously discovered fundamental immunological impairments of alveolar macrophages in COPD. Alveolar macrophages of adults with COPD had impaired inflammatory cytokine responsiveness to non-typeable Haemophilus influenzae (NTHI) outer membrane protein (OMP) P6, a Toll-like receptor 2 (TLR2) ligand and to lipooligosaccharide, a TLR4 ligand, suggesting impaired TLR signalling in COPD alveolar macrophages.4 Additionally, COPD alveolar macrophages had diminished phagocytosis of NTHI, suggesting wide-ranging intracellular signalling dysfunction.5 These findings support a potential role for TLR signalling in COPD-associated pathology. Interactions between human macrophages and respiratory pathogens are critical to the pathogenesis of COPD, and failure to evoke a strong proinflammatory alveolar macrophage response may permit evasion of immune clearance of respiratory pathogens, perpetuating infection.
Each bacterial species that causes infection in COPD displays unique dynamics of colonisation and interaction with human cells. NTHI, the most common cause of COPD exacerbations, exhibits heterogeneous patterns of colonisation with high turnover of strains interspersed with long-term persistence and induces strain-specific immunological responses.6 ,7 Moraxella catarrhalis, the second most common cause of COPD exacerbations, has shorter periods of colonisation.8 ,9 While NTHI and M catarrhalis are almost exclusively mucosal pathogens,7 ,9 Streptococcus pneumoniae causes invasive disease and community-acquired pneumonia, as well as mucosal pathology in COPD.10 ,11 Each pathogen presents a unique set of potential immunological triggers for different host pathways.
Our previous investigations were limited to study of NTHI and did not determine whether defective immune responsiveness discriminated among respiratory pathogens.4 ,5 Notably, these studies did not address the link between impaired alveolar macrophage innate defence and COPD exacerbations. Immunological links with exacerbations have been described. Diminished peripheral T lymphocyte proliferative responses to NTHI OMP P6, as well as sputum IL-6 and IL-8 induction, are associated with exacerbations.12–14 Moreover, increased risk of COPD exacerbation is associated with acquisition of new respiratory strains of NTHI.15 However, the underlying immunological triggers have not been explained. We hypothesised that dysfunctional innate immune responses in COPD, specifically impaired proinflammatory cytokine and TLR induction, are associated with COPD exacerbations and performed experiments to test this hypothesis.
Materials and methods
Subject recruitment
All procedures received VAWNY and University at Buffalo Institutional Review Board approval. After informed consent, volunteers were screened by spirometry (details provided in online supplement). Participants with stable COPD were enrolled and underwent bronchoscopy. All were followed prospectively for occurrence of exacerbations by bimonthly telephone contacts and 6-month and 12-month clinic visits. Participants experiencing at least one exacerbation requiring healthcare use over the study period were considered ‘exacerbation-prone’. Those without a defined exacerbation were labelled ‘non-exacerbation-prone’. Strict inclusion, exclusion and exacerbation criteria are detailed in the online supplement.
Purification of human alveolar macrophages
Ninety-six participants underwent right middle lobe bronchoalveolar lavage (BAL) to obtain alveolar macrophages.4 ,5 Follow-up evaluation was done 24 h postbronchoscopy. Alveolar macrophage purification is detailed in the online supplement.
Cytokine induction
-
Macrophage–bacteria interaction: Three distinct respiratory pathogens, obtained during COPD exacerbations, were used: NTHI 11P6H1; S pneumoniae 25P55S1 and M catarrhalis 6P29B1. Each donor's alveolar macrophages were inoculated with each strain (MOI- alveolar macrophage:bacteria=1:200). Supernatants were harvested (3 h) for IL-8 and TNF-α analysis. Details of controls, bacterial strains and dose–response curves are provided in the online supplement.
-
Macrophage–TLR ligand interaction: Alveolar macrophages were incubated with purified TLR2 (tripalmitoyl cysteine (Pam3Cys); EMC Microcollections GmbH, Tuebingen, Germany) and TLR4 (Escherichia coli K235 lipopolysaccharide (LPS); Sigma Chemical Co.) ligands (1 µg/mL; 3 h), to determine responses of alveolar macrophages to TLR2 and TLR4 ligands. Supernatants were harvested for IL-8 and TNF-α analysis. Dose–response and time–response curves are provided in the online supplement.
-
Cytokine analysis: IL-8 and TNF-α concentrations were determined by multianalyte microsphere flow cytometry.3 ,16 Lower limits of detection were 6.2 and 3.6 pg/mL, respectively. Concentrations were corrected for inter-experiment differences in macrophage numbers.
TLR expression
-
Immunostaining: Alveolar macrophages were adhered on chamber slides (Nunc Inc., Naperville, Illinois, USA). Following co-incubation (1 h) with each bacteria,5 adherent macrophages were immunostained for TLR2 and TLR4 expression, detailed in the online supplement.
-
Analysis: A minimum of five representative fields (25 cells each), for each condition for each participant, were captured digitally. Images were analysed (Image-Pro Plus V.6.3, Media Cybernetics Inc., Bethesda MD). Parameters were set to exclude background fluorescence. Fluorescent intensities were determined for each alveolar macrophage; mean fluorescent intensities (MFI) of TLR2 and TLR4 expression were averaged for each participant.
NF-κB activation
Alveolar macrophages were incubated with each bacteria or buffer alone. Nuclear protein was quantitated to obtain 20 µg nuclear protein per condition.17 Colorimetric assay for activation of NF-κB transcription factors was performed (Active Motif, Carlsbad, California, USA), detailed in the online supplement. Results are given as fold increase from untreated macrophages.
Statistical analysis
Demographic data were normally distributed and analysed by unpaired t test for continuous variables and χ2 test for nominal variables (table 1). Cytokine induction and TLR expression data between groups were not normally distributed and are reported as median (IQR) compared by Mann–Whitney U rank tests. Group differences were confirmed with univariate logistic regression and indicated age, FEV1(L) and FEV1(%predicted) to have p<1.0. Since FEV1(L) and FEV1(%predicted) are co-linear, only FEV1 was used for multivariate analysis. Multivariate logistic regression models in which age and FEV1 were added to each significant macrophage function tested were constructed to ascertain whether differences in macrophage function between exacerbators and non-exacerbators were still seen. For all comparisons, p≤0.05 was considered significant.
Demographic characteristics of participants
Results
Demographics of participants
Of 96 recruited volunteers meeting inclusion criteria for bronchoscopy for this study, eight were lost to follow-up. To determine the relationship of alveolar macrophage dysfunction with COPD exacerbations, the 88 remaining participants were prospectively identified as exacerbation-prone (n=29) and non-exacerbation-prone (n=59), as detailed earlier. Age (mean years±SEM) was not significantly different between exacerbation-prone and non-exacerbation-prone COPD subjects (62.0±1.7 vs 58.1±1.3; p=0.14). Participants included 66 men and 22 women. Demographic data are presented in table 1.
Exacerbation-prone COPD subjects had significantly worse spirometric parameters (FEV1; FEV1,%predicted) than did non-exacerbators. Therefore, regression analyses were used to determine the independent impact of group disparities in spirometric measures on immunological outcomes.
The numbers of alveolar macrophages obtained by bronchoscopy (means ×105±SEM) were 325.5±84.5 for COPD exacerbators and 488.1±64.1 for COPD non-exacerbators (p=0.14). No participants were taking systemic steroids or antibiotics. Fifty-four participants (59.3% of non-exacerbators; 65.5% of exacerbators) were taking bronchoactive medications, including inhaled corticosteroids in 17 (18.6% of non-exacerbators; 20.1% of exacerbators), β agonists in 52 (57.6% of non-exacerbators; 62.8% of exacerbators), anticholinergic medications in 34 (33.9% of non-exacerbators; 48.3% of exacerbators). For each intergroup comparison, there were no statistically significant differences in medication use.
Bacterial induction of cytokines
To determine the relative immune responsiveness of alveolar macrophages of each group to each respiratory pathogen, alveolar macrophages were incubated with NTHI, M catarrhalis and S pneumoniae and supernatants were assayed for IL-8 (figure 1) and TNF-α (figure 2). Data are expressed as median pg/mL (IQR). Baseline values (buffer-treated cells) for IL-8 were 1025 (2740) and 580 (2192) (p=0.32) and for TNF-α were 0 (22) and 0 (0.52) (p=0.36) for non-exacerbation-prone and exacerbation-prone COPD participants, respectively.
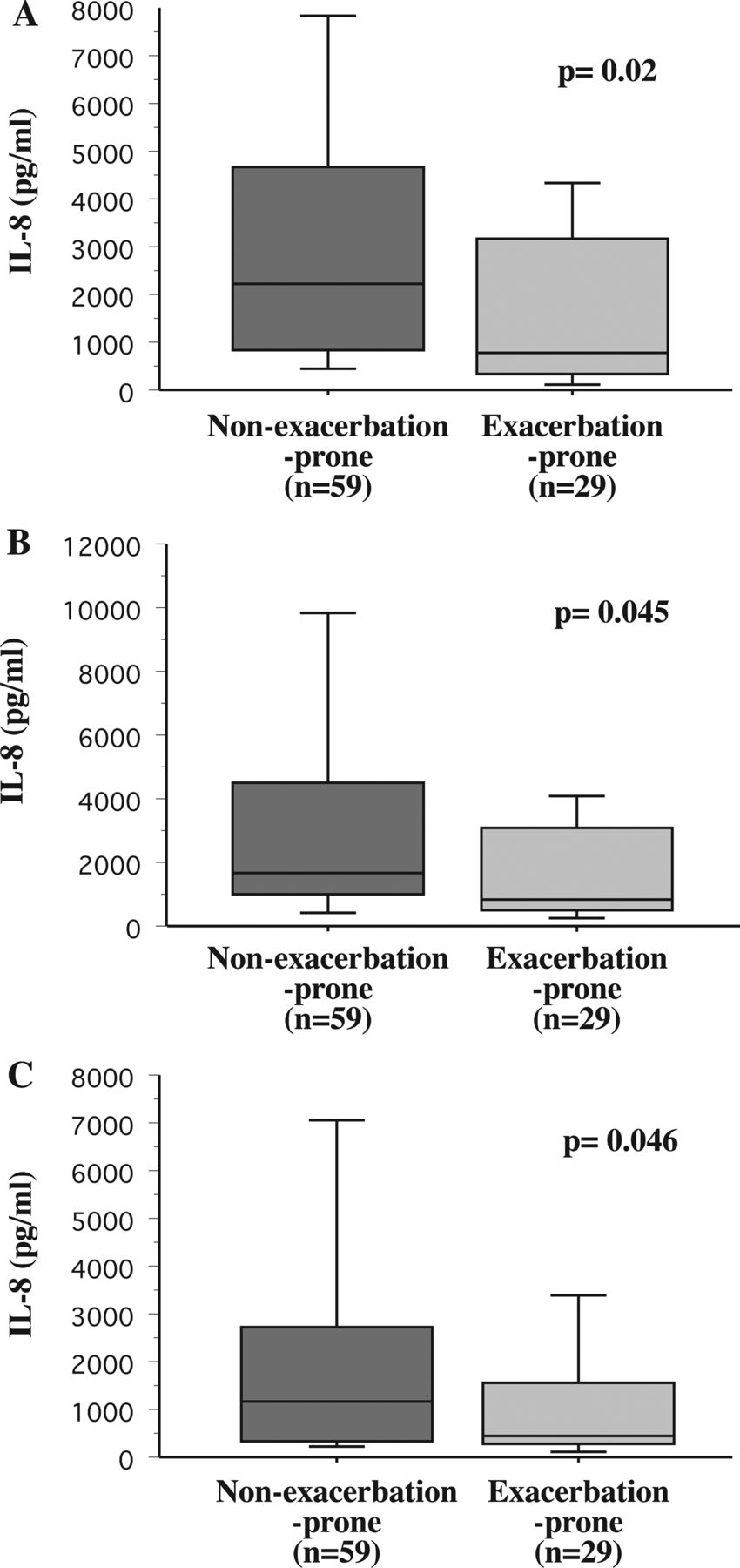
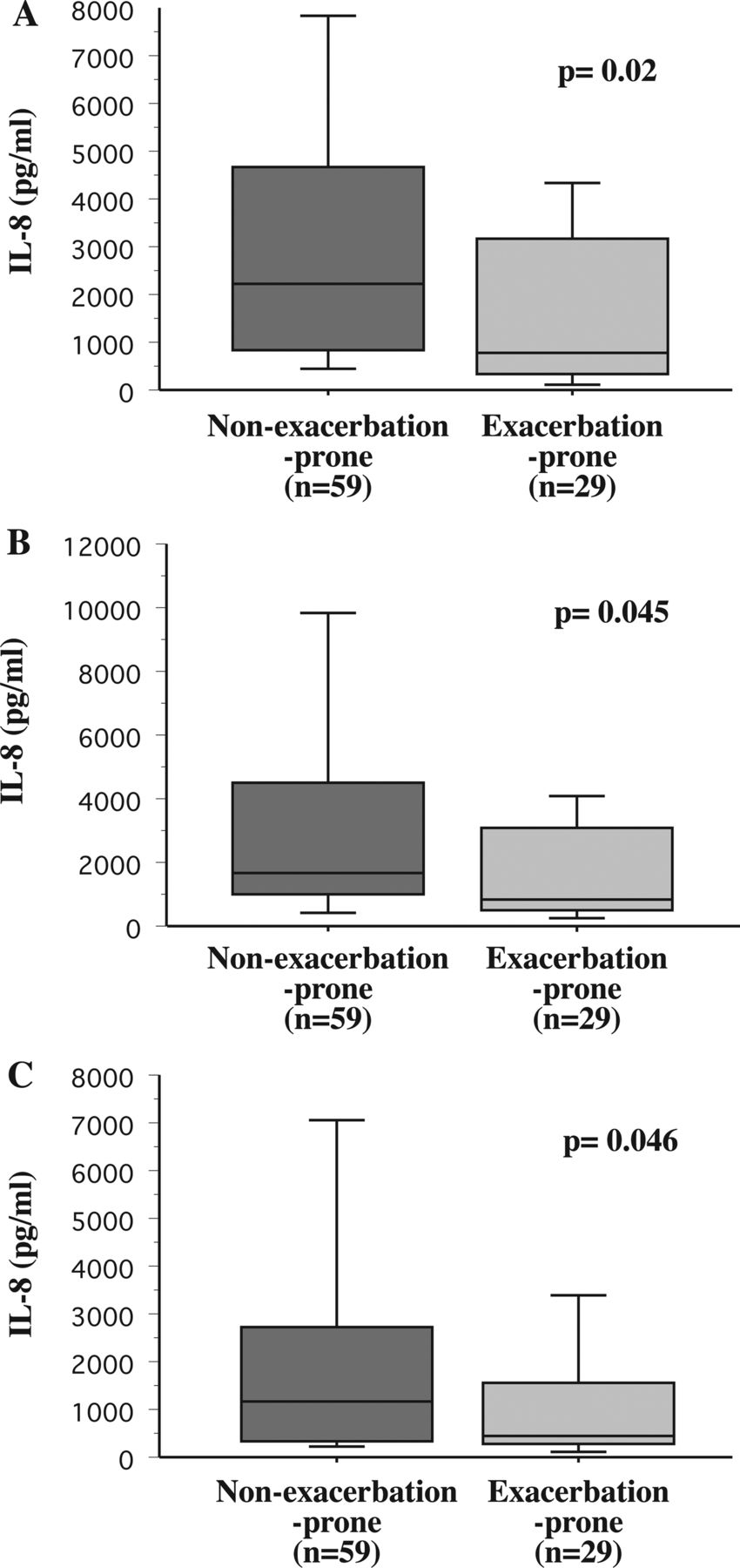
Bacterial induction of human alveolar macrophage IL-8 and COPD exacerbations. Alveolar macrophages were obtained from COPD participants who were identified prospectively over the course of the study as non-exacerbation-prone (dark grey shading) and exacerbation-prone (light grey shading). Cells were incubated with non-typeable Haemophilus influenzae 11P6H1 (A), Moraxella catarrhalis 6P29B1 (B) and Streptococcus pneumoniae 25P55S1 (C), and supernatant IL-8 concentrations were measured. Results are shown as box plots for each group. Each box encompasses the 25–75th IQR, with the horizontal line in each box representing median values. Each vertical bar encompasses the 10–90th centile ranges. Data correspond with the numeric values given in ‘Results’. Statistical comparison of groups was performed by Mann–Whitney U rank test.

Bacterial induction of human alveolar macrophage TNF-α and COPD exacerbations. Alveolar macrophages were obtained from COPD non-exacerbation-prone (dark grey shading) and exacerbation-prone (light grey shading), incubated with non-typeable Haemophilus influenzae (A) and Moraxella catarrhalis (B), and supernatant TNF-α concentrations were measured. Results are shown as box plots for each group. Data are represented by box plots for each group, as detailed in figure 1, and correspond with the values given in ‘Results’. Alveolar macrophages produced negligible concentrations of TNF-α in response to Streptococcus pneumoniae (not shown).
Alveolar macrophage cytokine responsiveness and COPD exacerbations
IL-8: To determine the relationship of alveolar macrophage dysfunction with COPD exacerbations, alveolar macrophages of exacerbation-prone and non-exacerbation-prone participants were incubated with bacterial pathogens. Alveolar macrophages of exacerbation-prone COPD individuals were more refractory (p=0.02) to NTHI-induction of IL-8 (median pg/mL (IQR)) than were their non-exacerbation-prone counterparts (775.0 (2814) vs 2365 (4250.5); figure 1A). Alveolar macrophage responsiveness to M catarrhalis-induction of IL-8 (870 (2586) vs 1710 (3383.8); p=0.045; figure 1B) and to S pneumoniae-induction of IL-8 (460 (1319) vs 1165 (2137.4); p=0.046; figure 1C) was also diminished among exacerbation-prone COPD participants. Thus, clear association exists between occurrence of prospectively measured COPD exacerbations and alveolar macrophage IL-8 responsiveness to respiratory bacterial triggers. Analysis of stimulated values, corrected for baseline cytokine expression, is detailed in the online supplement, further corroborating refractoriness of alveolar macrophages of exacerbation-prone participants.
TNF-α: Alveolar macrophages of exacerbation-prone COPD individuals were also more refractory (p=0.046) to NTHI-induction of TNF-α than were their non-exacerbation-prone counterparts (0 (4.3) vs 0 (19.2); figure 2A). Although differences were not statistically significant, M catarrhalis-induction of TNF-α was also diminished for alveolar macrophages of exacerbation-prone participants (7.0 (112.4) vs 16.1 (515.0); figure 2B) (p=0.08). S pneumoniae elicited only negligible TNF-α concentrations (not shown).
TLR ligand cytokine induction and COPD exacerbations
IL-8: To determine the role of alveolar macrophage TLR signalling in COPD exacerbations, alveolar macrophages of exacerbation-prone and non-exacerbation-prone participants were incubated with purified TLR2 (Pam3Cys) and TLR4 (LPS) ligands to measure induction of IL-8. TLR4-mediated IL-8 induction of alveolar macrophages of exacerbation-prone COPD individuals was impaired (p=0.028) compared with responses of non-exacerbators (500 (1361) vs 1220 (1617); figure 3A). Although differences in TLR2-mediated IL-8 induction did not achieve statistical significance (p=0.074), results showed a trend towards diminished responses among alveolar macrophages of exacerbation-prone donors (640 (1440) vs 1160 (2179); figure 3B).

Toll-like receptor 2 (TLR2) and TLR4 ligand induction of human alveolar macrophage IL-8. Alveolar macrophages, obtained from COPD non-exacerbation-prone (dark grey shading) and exacerbation-prone (light grey shading) participants, were incubated with the TLR4 ligand lipopolysaccharide (LPS, A) and with the TLR2 ligand tripalmitoyl cysteine (Pam3Cys, B), and supernatant IL-8 concentration was measured. Data are represented by box plots for each group, as detailed in figure 1, and correspond with the numeric values given in ‘Results’.
TNF-α: Alveolar macrophages of exacerbation-prone COPD individuals were also more refractory (p=0.015) to TLR4-mediated (4.7 (20.5) vs 18.8 (91.9); figure 4A) and to TLR2-mediated (21.3 (60.6) vs 53.5 (119.8)); figure 4B) induction of TNF-α (p=0.049) than were their non-exacerbation-prone counterparts.

Toll-like receptor 2 (TLR2) and TLR4 ligand induction of human alveolar macrophage TNF-α. Alveolar macrophages, obtained from COPD non-exacerbation-prone (dark grey shading) and exacerbation-prone (light grey shading) participants, were incubated with lipopolysaccharide (A) and with Pam3Cys (B), and supernatant TNF-α concentration was measured. Data are represented by box plots for each group, as detailed in figure 1, and correspond with the values given in ‘Results’.
NF-κB activation
To determine whether impaired immunological responsiveness of exacerbation-prone COPD alveolar macrophages is mediated through transcriptional regulator NF-κB, nuclear extracts of alveolar macrophages incubated with each bacteria were analysed. Comparison was made with same-donor baseline values and is expressed as the fold increase for each. Fold increase of nuclear NF-κB for exacerbation-prone vs non-exacerbation-prone COPD was 4.1 (3.5) vs 10.1 (5.9) in response to NTHI (p=0.02); 5.8 (2.5) vs 17.9 (22.0) in response to M catarrhalis (p=0.04) and 3.0 (5.1) vs 16.1 (16.1) in response to S pneumoniae (p=0.02).
TLR expression and COPD exacerbations
To determine the role of microbial regulation of alveolar macrophage TLR expression, MFIs of bacteria-induced TLR2 and TLR4 were analysed (table 2). Alveolar macrophages of active smokers exhibited excessive autofluorescence, making results difficult to interpret reliably. Therefore, analysis for these specific studies was limited to ex-smokers. Baseline MFI values (buffer-treated cells) for TLR2 were 7.4 (3.7) and 7.7 (6.0) (p=0.53) and for TLR4 were 8.9 (4.3) and 7.6 (4.4) (p=0.26) for non-exacerbation-prone and exacerbation-prone participants, respectively. However, M catarrhalis-induced (14.0 (6.3) vs 17.2 (4.2); figure 5, top row) and S pneumoniae-induced (16.1 (4.7) vs 19.8 (5.3)); figure 5, middle row) TLR2 expression was significantly diminished in exacerbation-prone donors (p=0.04 for each). NTHI-induced TLR2 expression did not differ between the two groups (figure 5A; p=0.89). Induced TLR4 expression was no different between alveolar macrophages of exacerbation-prone and non-exacerbation-prone participants in response to all three bacterial pathogens (table 2).
Bacterial induction of alveolar macrophage TLR2 and TLR4 expression.

Bacterial induction of human alveolar macrophage Toll-like receptor 2 (TLR2) expression. Alveolar macrophages, obtained from non-exacerbation-prone (left) and exacerbation-prone (right) COPD participants, were incubated with Moraxella catarrhalis (top row), Streptococcus pneumoniae (middle row) and non-typeable Haemophilus influenzae (bottom row), and TLR2 expression was measured. Images shown are from one participant of each group. Complete mean fluorescent intensities for all participants of each group are given in table 2.
To establish the relationship between bacteria-induced TLR expression and bacterial induction of IL-8, regression analyses were performed (figures 6 and 7) and confirmed by Spearman correlation. Clear association exists between NTHI-induced IL-8 and expression of both TLR2 (figure 6A; p=0.04; r=0.432) and TLR4 expression (figure 7A; p=0.05; r=0.398). M catarrhalis-induced IL-8 also correlated with induced TLR2 (figure 6B; p=0.0003; r=0.671) and TLR4 expression (figure 7B; p=0.007; r=0.54). S pneumoniae-induced IL-8 also had a strong relationship with induced TLR2 (figure 6C; p<0.001; r=0.778) and TLR4 (figure 7C; p=0.006; r=0.555) expression. Thus, strong association exists between induction of IL-8 for each bacteria and TLR expression.

Bacterial induction of IL-8 and Toll-like receptor 2 (TLR2) expression. Correlation of induction of IL-8 by non-typeable Haemophilus influenzae (A), Moraxella catarrhalis (B) and Streptococcus pneumoniae (C) from alveolar macrophages of non-exacerbation-prone and exacerbation-prone ex-smoking COPD participants and TLR2 expression is shown. Statistical correlation was determined by regression analyses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bacterial induction of IL-8 and Toll-like receptor 4 (TLR4) expression. Correlation of induction of IL-8 by non-typeable Haemophilus influenzae (A), Moraxella catarrhalis (B) and Streptococcus pneumoniae (C) from alveolar macrophages of non-exacerbation-prone and exacerbation-prone ex-smoking COPD participants and TLR4 expression is shown. Statistical correlation was determined by regression analyses.
Multivariate regression analyses
Because demographic group differences existed in age and FEV1 (table 1), we created multivariate logistic regression models to determine whether intergroup differences in macrophage response were independent of demographic differences. Since cytokine induction data were not normally distributed, base 10 logarithmic values were used for the regression model. Tables with complete data (p values, ORs and 95% confidence indices) are included in the online supplement. In each of these models, macrophage IL-8 induction and TLR2 induction remained significantly different between exacerbators and non-exacerbators. All macrophage immunological responses that were significantly different as individual variables between the two groups remained so in the multivariate logistic regression, except for Pam3Cys-induced TNF-α. Thus, demographic variables had minimal statistical effect on overall alveolar macrophage cytokine and TLR expression results.
Discussion
This is the first study to demonstrate distinct correlation of alveolar macrophage dysfunction with clinical predilection towards COPD exacerbations. Furthermore, this association occurred in response to NTHI, M catarrhalis, and S pneumoniae, the three most common respiratory pathogens in COPD.19 An evoked alveolar macrophage inflammatory response is critical for innate immune-mediated bacterial clearance. Occurrence of bacterial exacerbations is primarily driven by acquisition of new bacterial pathogens in the lower respiratory tract.2 Increased lower airway inflammation has been linked to increased respiratory symptoms in COPD leading to clinical exacerbations.20 With the introduction of new strains of respiratory pathogens and impaired macrophage clearance, bacteria persist. Failure to clear respiratory bacteria may promote exacerbations and require adaptive immune mechanisms for pathogen clearance.
We previously discovered that alveolar macrophages of adults with COPD had impaired inflammatory cytokine responsiveness to NTHI OMP P6 and LOS, suggesting impaired signalling through TLR2 and TLR4 pathways.4 While TLR4 signalling plays a role in cigarette smoke-mediated inflammation, there is limited investigation into TLR–microbial interactions in COPD exacerbations.21 Studies in murine models support a role for TLRs in NTHI clearance from lung tissue.22 ,23 This prompted us to evaluate not only alveolar macrophage signalling though TLRs, but also microbial regulation of TLR expression in exacerbation-prone COPD individuals. The refractoriness of exacerbation-prone COPD alveolar macrophages to specific TLR2 and TLR4 ligands, by both IL-8 and TNF-α responsiveness in this study, demonstrates impaired TLR2 and TLR4 signalling among those prone to COPD exacerbations. Furthermore, diminished TLR2-mediated induction of TNF-α, as well as TLR4 induction of IL-8 and TNF-α, supports broad impairment of alveolar macrophage TLR signalling among COPD exacerbation-prone individuals, including impaired mediation of signalling through NF-κB, a pivotal event in TLR-mediated activation of innate immune response.24 The relative contributions of genetic factors and smoking to this impairment warrant further investigation. Our results suggest that determinants of TLR responsiveness provide some degree of explanation for the wide variation in exacerbation frequency.
Since colonisation with respiratory pathogens might serve as a macrophage priming mechanism, we cultured each BAL for respiratory bacteria. NTHI was retrieved from BAL of only one exacerbation-prone and two non-exacerbation-prone participants. M catarrhalis and S pneumoniae were not recovered from any samples. This likely reflects our selection criteria for individuals who were asymptomatic at the time of bronchoscopy.
Several mechanisms could mediate impaired TLR responses. The induction of TLR2 molecules on alveolar macrophages in response to M catarrhalis and S pneumoniae was associated with a propensity for exacerbations among COPD ex-smokers. Moreover, induced TLR expression was strongly associated with bacterial induction of IL-8. Collectively, these findings support a role for impaired alveolar macrophage TLR expression and signalling through bacterial cytokine induction in exacerbation-prone COPD. Related studies have begun to explore induced TLR expression as a component of immune dysfunction in COPD, but have focused on sputum neutrophils and blood monocyte-derived macrophages.25 ,26 In fact, TLR2 expression in sputum neutrophils may be subject to different regulation in COPD.25 Moreover, induced TLR2 expression in alveolar macrophages was noted to be reduced among small numbers of individuals with COPD, although no correlation was sought with exacerbations in this study.25 Study of TLR expression and potential regulation by bacterial products and cigarette smoke in alveolar macrophages and in COPD has been investigated.26 ,27 However, the relationship of microbial regulation of alveolar macrophage TLR expression to COPD exacerbations has not been previously explored. The strong association between induction of IL-8 for each bacteria and TLR expression supports a relationship, although further studies will be needed to establish an unambiguous link.
Exacerbations contribute substantially to morbidity and mortality in COPD.3 In fact, frequency of exacerbations is associated with shifts in proinflammatory cytokines, underscoring the importance of fundamental immune regulation.28 ,29 Increased IL-8 in BAL fluid has been a distinguishing marker of COPD in active smokers.30 ,31 Thus, it is intriguing that alveolar macrophages of exacerbation-prone individuals with COPD were refractory to induction of IL-8 compared with non-exacerbation-prone COPD. This suggests that alveolar macrophage IL-8 induction in exacerbation-prone COPD might be subject to a different regulation than their non-exacerbation-prone counterparts and that IL-8 in BAL fluid of COPD active smokers may be largely a product of neutrophils and epithelial cells.32
While our results are definitive, there are inherent limitations to our study. Exacerbation frequency increases with worsening COPD, and the need for bronchoscopy limited our investigation to mild-to-moderate COPD.33 Although the correlation of induced TLR expression for exacerbations among ex-smokers was definitive, our study of induced TLR expression does not address this immunological feature for exacerbations among COPD active smokers. Finally, while diminished IL-8 and TNF-α responsiveness of alveolar macrophages correlated with a propensity towards exacerbations, the duration and size of our study did not allow us to distinguish individuals with more frequent, from those with less frequent, exacerbations.
The present study advances our understanding of innate immune dysfunction in COPD, revealing distinct impairment of alveolar macrophages among individuals prone to exacerbations. Notably, clinical predilection towards COPD exacerbations strongly correlates with refractoriness of alveolar macrophage cytokine response to respiratory pathogens. Impaired TLR2 and TLR4 signalling and bacterial induction of TLR expression comprise an integral part of this dysfunction. Studies to address the underlying cellular mechanisms of these processes will be directed towards therapeutic strategies to improve macrophage responsiveness and reduce exacerbations in COPD. Activation of the nuclear erythroid-related factor 2 (Nrf2) pathway resulted in enhanced alveolar macrophage clearance of bacteria, suggesting a potential role in exacerbation-prone COPD.34 Collectively, our findings support a dynamic immunological interaction between alveolar macrophages and respiratory pathogens as a component of predilection towards COPD exacerbations.
Acknowledgments
The authors are grateful for the scientific advice offered by Timothy F Murphy. This manuscript is dedicated to the memory to our colleague, Jane Maloney, without whom this work would not have been possible.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
Footnotes
-
Contributors CSB and SS provided conception and design of project, data analysis and interpretation of data. RLK did all cell biology laboratory work. EE performed acquisition of all clinical data. PKW performed multianalyte flow cytometry and cytokine data analysis. HM and RD performed fluorescent analysis.
-
Funding This work was supported by research grant R01HL082561-01 (CSB, SS) from the National Institutes of Health and with support from the Department of Veterans Affairs.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval VA Western New York and University at Buffalo Institutional Review Boards.
-
Provenance and peer review Not commissioned; externally peer reviewed.