Article Text

Abstract
Epithelial–mesenchymal transition (EMT) is a process when epithelial cells gradually transform into mesenchymal-like cells losing their epithelial functionality and characteristics. EMT is thought to be involved in the pathogenesis of numerous lung diseases ranging from developmental disorders, fibrotic tissue remodelling to lung cancer. The most important question—namely what is the importance and contribution of EMT in the pathogenesis of several chronic lung conditions (asthma, COPD, bronchiolitis obliterans syndrome and lung fibrosis)—is currently intensely debated. This review gives a brief insight into the mechanism and assessment methods of EMT in various pulmonary diseases and summarises the recent literature highlighting the controversial experimental data and conclusions.
Statistics from Altmetric.com
What is epithelial–mesenchymal transition?
Epithelial–mesenchymal transition (EMT) is a process in which epithelial cells gradually acquire a mesenchymal (fibroblast-like) cell phenotype. EMT has been shown to be essential for embryonic development, gastrulation, and the development of the neural crest, heart and other organs. EMT is also implicated in tissue repair, organ fibrosis and cancer progression. EMT is associated with the progression of many type of tumours and is involved in their metastasis and treatment resistance.1 ,2
Cellular changes in EMT
During EMT, epithelial cells undergo profound changes which can be categorised into four broad cellular functions: changes in transcriptional regulation; changes in cytoskeleton and motility; changes in cell adhesion; and changes in the synthesis of extracellular matrix (ECM) components.
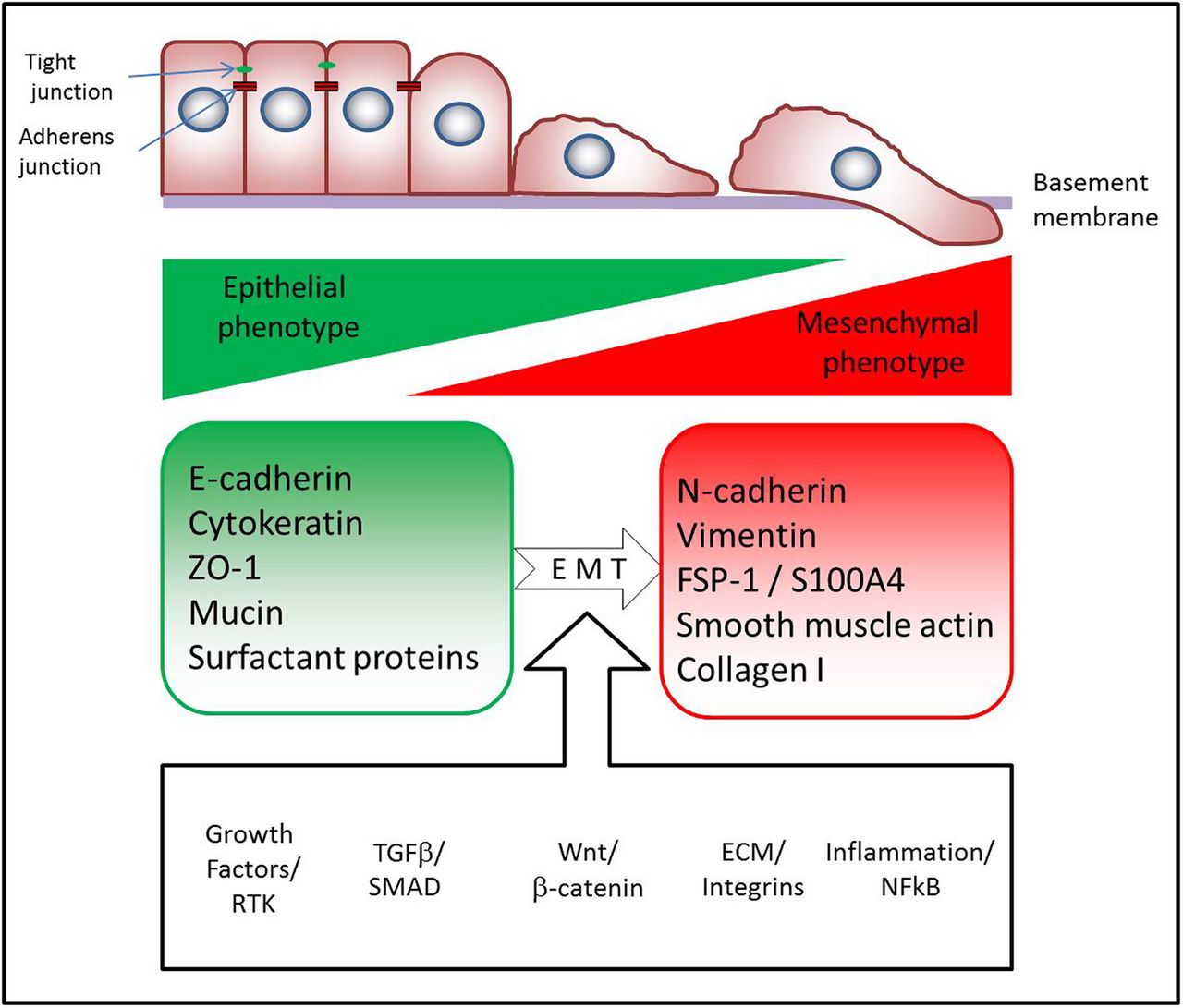
Several transcription factors such as Snail, Slug, ZEB-1, ZEB-2, Twist, β-catenin and Tcf/LEF have been identified as key regulators of EMT, and are the ‘master switches’ important for cell reprogramming (figure 1).2–5
{kind=link}
The most important features of epithelial–mesenchymal transition (EMT). Multiple pathways have been found to trigger EMT: tyrosine kinase receptors (epidermal growth factor, fibroblast growth factor, connective tissue growth factor, platelet-derived growth factor, insulin-like growth factor, etc), integrins, Wnt, nuclear factor (NF)-κB and transforming growth factor β (TGF-β) pathways. A common feature of these pathways is that they activate ‘master transcription factors’ (Snail, Slug, Zeb-1, Twist, etc) which switch on the EMT programme in epithelial cells, namely, downregulating E-cadherin expression and other genes linked to the epithelial phenotype and activating the transcription of genes associated with the mesenchymal phenotype. Canonical TGF-β/SMAD signalling is perhaps the best characterised signalling pathway which contributes to fibrosis (for a recent review, see Fernandez et al90). Activation of canonical Wnt signalling and β catenin makes epithelial cells susceptible to EMT caused by TGF-β, by inhibiting the growth arrest which is induced by TGF-β. The extracellular matrix (ECM) also modulates TGF-β signalling through integrin signalling contributing to EMT. Inflammatory cytokines activating NF-κB are thought to further accentuate fibrosis and EMT in the lung.
Cytokeratins are the characteristic intermediate filaments in epithelial cells. During EMT cells are reprogrammed to express vimentin instead.2 ,5 Other cytoskeletal changes involve de novo expression of fibroblast specific protein (FSP)-1 (also called S100A4) in the epithelium, which has been associated with increased migratory capacity, a more invasive phenotype in tumours and also worse prognosis in lung cancer.6 α-Smooth muscle actin (α-SMA) is also a frequently assessed marker in EMT. Expression of α-SMA is characteristic for myofibroblasts which are laying down an excessive amount of ECM and thus are responsible for tissue remodelling and fibrosis (figure 1).7
Early molecular signs of EMT involve the dissociation of cell–cell junctions induced by downregulation and/or disassembly of junctional components, such as occludins, ZO-1, claudins and E-cadherin.2 ,5 The gradual loss of E-cadherin—considered a hallmark of EMT—is accompanied by the simultaneous gain of N-cadherin, a mesenchymal marker. This change is commonly termed the ‘cadherin switch’ (figure 1).5
Cells undergoing EMT also begin to synthesise ECM components like fibronectin and collagen type 1.8 ,9
Assessment of EMT in lung tissue
EMT is thought to be a gradual process, meaning that there are intermediate cellular phenotypes during the transition. An epithelial cell undergoing EMT will express a set of different marker genes during this process. Since changes in gene expression have also been detected during injury, activation or dedifferentiation of epithelial cells, it is obviously rather challenging to identify with certainty epithelial cells undergoing EMT in complex pathological conditions.
It is well established that during the physiological repair process following epithelial injury, loosening of epithelial–epithelial cell contacts (eg, tight junctions and adherent junctions), alteration of cell–ECM contacts, and cytoskeletal activation (migration) and morphological changes (spreading) occur during wound repair.10 These phenotypic changes are thought to be mediated by EMT transcription factors.11 However, we have to emphasise that during physiological wound repair mesenchymal markers expressed by epithelial cells do not necessarily mean transdifferentiation to fibroblasts or myofibroblasts. Although acquiring mesenchymal features and molecular markers, migrating epithelial cells in wound repair do not downregulate E-cadherin and tight junction proteins; there is rather an intercellular redistribution of these proteins.11–13
There are numerous articles demonstrating that epithelial cells coexpress mesenchymal and epithelial markers in various lung diseases in humans.14–20 Other methods commonly used to identify EMT are to show the activation of signalling pathways and transcription factors thought to regulate EMT. These mechanisms—for example, the activation of transcription factors of the Snail family and the consequent downregulation of E-cadherin—show striking similarities between fibrosis, developmental programmes involving EMT and carcinogenesis, suggesting a common molecular regulatory background is present.21–23 However, these experimental methods only provide a ‘snapshot’ of a process stretched over a period of time and thus are criticised for not being suitable to follow a dynamic process like EMT. To solve this ongoing controversy there is a need for methods which are able to track the transdifferentiation process more reliably, like intravital microscopy or cell fate mapping.
Currently there are no publications available describing the use of intravital microscopy to evaluate EMT of lung epithelial cells in real time. Faulkner et al24 found that kidney interstitial fibroblasts do not derive from tubular epithelial cells in renal fibrosis and it was described that activation of intrinsic transforming growth factor β (TGF-β) signalling is essential in blood-borne metastasis formation in breast cancer cells using in vivo imaging techniques.25
However, the most important question remaining is whether EMT causally contributes to lung disease and, if yes, to what extent? Lung conditions with chronic inflammation and persistent injury result in scarring, fibrosis and tissue remodelling, leading to a decrease of lung function, respiratory failure and death. Recently, experiments using genetic cell fate tracking of lung epithelial cells were performed to address this issue in mouse models of lung fibrosis9 ,26 ,27 and allergic asthma.28 These studies found that approximately 30–50% of murine lung fibroblasts in lung fibrosis and allergic asthma were derived from epithelial cells which had undergone EMT as identified by genetic tagging. In these models, β galactosidase was used as a genetic label.9 ,26 ,27 Criticism of this method highlighted that the enzymatic reaction using X-gal and β galactosidase does not allow high-resolution confocal imaging contrary to fluorescent tags. Because of this, the borders between neighbouring cells are obscure and the possibility of false-positive identification of cells cannot be excluded.29 Additionally, these lineage-tracking studies9 ,26 ,27 used transgenic mice in which the recombination was driven by a 3.7 kb fragment of the human surfactant protein C (Sftpc) promoter,30 which is somewhat different from the endogenous murine Sftpc promoter which drives the recombination in other animal models. 29 Also in vitro culture of alveolar epithelial cells (AECs) after isolation may result in experimental artefacts, which raises doubt as to whether EMT actually contributes to in vivo fibroblast formation.8 ,9 ,26
Recently published data have challenged the idea that a significant fraction of myofibroblasts in the diseased lung derive from epithelial cells in animal models29 ,31 and humans.32 These contradictive results from different laboratories have resulted in an intensive debate on lung fibrosis, and kidney33 ,34 and liver fibrosis.35 Rock et al used a transgenic mouse strain with a fluorescent protein tag (Tomato) under the control of the endogenous Sftpc promoter. These experiments suggests that AECs do not transdifferentiate into myofibroblasts in large numbers in the bleomycin lung injury model.29 A recently published study in mice using multiple genetic tags for cell fate tracking found that myofibroblasts in unilateral urethral obstruction (UUO) kidney fibrosis derive from proliferation of local precursors (50%), bone-marrow-derived fibrocytes (35%), endothelial-mesenchymal transition (10%) and EMT (5%).34 The study of LeBleu et al34 using the UUO model is a good example, highlighting the multiple origins of myofibroblasts. However, it is difficult to extrapolate from the UUO kidney fibrosis model to the lung as the organ function, the mechanism of parenchymal injury and consequent fibrotic changes are considerably different.
These controversial data illustrate the ongoing intensive scientific debate on the origin of myofibroblasts in fibrotic diseases and tissue remodelling. Elucidating this further is crucial to determine the direction of future research and identification of new therapeutic targets.
EMT in lung development and paediatric diseases
The first EMT event described occurs in gastrulation—the earliest stage of embryonic development. During this process, the primary mesenchyme is formed, giving the first distinction between epithelial and mesenchymal phenotypes. The epithelial and mesenchymal cell phenotypes are not irreversible, and during embryonic development, cells can convert between the epithelial and mesenchymal states through the process of EMT or mesenchymal-to-epithelial transition (MET).36 ,37 Sometimes, several rounds of EMT and MET are required to give a rise to specialised cell types needed for organ formation. During development, EMT provides flexibility in cell fate; however, the development of specialised tissues during embryogenesis requires additional epithelial cell plasticity that slightly differs from classical EMT. Epithelial cell plasticity is essential throughout the process of branching morphogenesis in numerous organs, including the lung.38 ,39 Lung-branching morphogenesis requires reciprocal signalling between the epithelium and surrounding mesenchyme that is mediated by coordinated activity of TGF-β/bone morphogenetic protein, Wnt, Sonic hedgehog, fibroblast growth factor (FGF) or retinoic acid signalling.40 Dysregulation of these signalling pathways is implicated in aberrant lung branching, alveologenesis and pulmonary vascular development, events critical for normal lung development. In addition to genetic factors, pre-natal and post-natal environmental exposures, including epigenetics, play a significant role in programming lung morphogenesis and development of paediatric lung disease.
Bronchopulmonary dysplasia (BPD) is a chronic lung disease that occurs in very premature infants and is characterised by impaired alveologenesis and vascular development. BPD develops as a result of injury or infection on a very immature lung. Interstitial fibrosis is recognised as a prominent feature of BPD as a consequence of repetitive lung injury. Deng and coworkers,41 using transgenic reporter mice, showed that lung fibroblasts in BPD could arise from bone-marrow-derived ATII cells that have undergone EMT. Noseda and colleagues42 ,43 revealed that expression of Notch1 or Notch4 in endothelial cells can cause transition to a mesenchymal phenotype via endothelial-to-mesenchymal transition, a process very similar to EMT. Constitutive Notch1 activation, together with inhibitor studies in lung organ cultures, identified a role for Notch signalling together with TGF-β in mesothelial EMT.44 Que et al45 have also shown that mesothelial cells covering the lung surface can migrate into the organ itself and give rise to various cell types, including mesenchymal cells, within the developing lung.
Taken together, these findings have important implications for our understanding of lung developmental defects; however, the casual impact of EMT to BPD has yet to be determined.
EMT in asthma
Airway remodelling comprises thickening of the basement membrane accompanied by sub-epithelial fibrosis, epithelial shedding and smooth muscle hypertrophy/hyperplasia.17 Under normal conditions, the damaged epithelium is able to repair itself quickly. However, constant damage inflicted by chronic inflammation in asthma results in a greatly reduced number of ciliated cells, leaving the basal membrane surface only partially covered with basal cells which are more resistant to apoptosis.17 Loss of E-cadherin as a hallmark of EMT in airway epithelial cells of patients with asthma is also described.14 ,46 ,47 Fibroblasts and myofibroblasts are responsible for the increased production of ECM proteins, for example, collagen and proteoglycans, and also serve as storage for cytokines and growth factors like TGF-β, which play important roles in fibrosis and the regulation of fibroblast–myofibroblast differentiation in inflamed airways.48 ,49
Recent experimental data indicate that T helper 2 dominant chronic inflammation leads to elevated TGF-β1 levels in airways via increased production of TGF-β by eosinophils and macrophages, in addition to fibroblasts, endothelial and smooth muscle cells. TGF-β is stored in the ECM and in turn induces EMT and sub-epithelial fibrosis.28
Concerning the pathogenesis of asthma, does EMT contribute to tissue remodelling and sub-epithelial fibrosis? Johnson and colleagues28 found evidence for this phenomenon in a house dust mite (HDM) allergen sensitised transgenic murine model. They demonstrated that β-galactosidase-tagged large airway epithelial cells gradually lose epithelial characteristics, gain expression of mesenchymal markers, for example, vimentin, and migrate into the sub-epithelial compartment.28 In vivo cell-fate tracking of genetically labelled epithelial cells provides evidence for epithelial cell transdifferentiation in this mouse model of chronic asthma. Other studies use primary human bronchial epithelial cell (hBEC) cultures to study the role of allergen sensitisation in EMT. HDM sensitisation of the airways synergises with TGF-β1 and results in the loss of E-cadherin expression,47 caveolin-1 internalisation and consequent loss of epithelial barrier function.46 Hackett et al14 investigated EMT using air–liquid interface cultures of hBECs isolated from patients with asthma and non-asthmatic controls. The authors found that basal cells of the stratified airway epithelium are the most susceptible to TGF-β1-induced EMT. Interestingly, they found no histological evidence for expression of mesenchymal markers in the bronchial epithelium of matched patients with asthma, rather an increased number of cytokeratin-5+ (KRT5+) positive cells, indicating a differentiation shift towards the basal epithelial cell phenotype.14 The results discussed above highlight the controversy and discrepancy between research data concerning EMT.
EMT in bronchiolitis obliterans syndrome
Bronchiolitis obliterans syndrome (BOS) is a major cause of allograft dysfunction in lung transplant recipients. Hodge et al15 found increased expression of mesenchymal proteins by large airway bronchial epithelial cells (BECs) in patients with BOS after lung transplantation to be a clear indicator of the presence of EMT. Ward et al50 found that S100A4 and matrix metalloproteinase (MMP)-2, MMP-7 and MMP-9 expression was present in the airway epithelium of patients who were stable following lung transplant without signs of BOS. Invasive capacity of BECs isolated from these patients was also shown. It is known that allograft infections pose a risk for BOS and has been shown to promote EMT in patient-derived small airway epithelial cells.51 Inadequate graft microvasculature and consequent graft ischaemia has emerged as a promoting factor for EMT in transplanted lungs.52 ,53 Intriguingly, an in vitro study suggests that commonly used immunosuppressive agents may promote EMT in BECs.54
EMT in COPD
COPD is accompanied by inflammation and tissue remodelling,55 and has been known to be strongly associated with lung cancer. A recent study found a robust genomic link between COPD, lung cancer and Hedgehog signalling, which is also implicated in tobacco-smoke-induced EMT.56 Tissue remodelling in COPD is characterised by emphysema, and small airway remodelling with peribronchiolar fibrosis. Recent investigations reported that nicotine and tobacco smoke induce EMT in BECs in a Wnt3a/β-catenin/TGF-β-dependent manner.57 Wang et al58 described that urokinase plasminogen activator receptor is overexpressed in the airway epithelium of patients with COPD and is involved in mediating the effects of tobacco smoke on EMT in BECs. Studies in mice chronically exposed to tobacco smoke showed increased levels of TGF-β1, connective tissue growth factor (CTGF) and platelet-derived growth factor (PDGF)-B in the treatment group.59 All these growth factors are known to induce EMT.2 ,60 ,61 Recent clinical investigations in patients with COPD18 ,20 showed that cells costaining for epithelial and mesenchymal markers are present in the large airways of asymptomatic smokers and in especially large numbers in current smokers. The authors showed that the reticular basement membrane (Rbm) in people who smoke and those with COPD is highly fragmented, with elongated spaces or cracks termed ‘clefts’, which is a hallmark of EMT in the airways.62 ,63 These clefts in the Rbm usually contained cells positive for mesenchymal markers S100A4, vimentin and MMP-9.20 Recent findings indicate that similar changes can also be observed in the small (<1 mm) airways of smokers.64
EMT in idiopathic pulmonary fibrosis
The histological finding characteristic for idiopathic pulmonary fibrosis (IPF) is usual interstitial pneumonia (UIP) with scattered α-SMA-positive fibroblastic foci (FF) in collagen-rich areas, alternating with normal lung areas. The most often used animal model for IPF is the single-dose bleomycin challenge, although it is known that several features of IPF—most importantly being a progressive and chronic disease—are poorly represented. Also, many drugs including corticosteroids proved to be effective in the bleomycin model but are ineffective in IPF.65 A recently published study suggests that a repetitive bleomycin injury model recapitulates several features of human IPF better than the conventional single-dose model. In this model the authors found that about 50% of S100A4+ fibroblasts are epithelial derived using genetic fate tracking.27 On the contrary, Rock et al29 found that epithelial cells do not contribute to fibroblast formation in the single-dose model.
Human immunohistochemistry studies on the role of EMT in the formation of myofibroblasts in IPF are controversial.66 Harada et al67 found myofibroblast cells in FF expressing epithelial markers TTF-1, cytokeratin and surfactant protein B. Chilosi et al68 identified migratory markers laminin5-γ2, heat shock protein 27 and fascin expression in epithelial cells clustering above FF in UIP samples with special ‘sandwich morphology’. On the contrary, Yamada et al32 did not find histological evidence for EMT in tissue samples from patients diagnosed with IPF (n=15) and non-specific interstitial pneumonia (n=12) using dual immunohistochemistry. In conclusion, the evidence that EMT is involved in the formation of activated myofibroblasts in pulmonary fibrosis remains controversial.
EMT in lung cancer
EMT is thought to contribute to cancer invasion and metastasis by allowing malignantly transformed epithelial cells to migrate, invade the surrounding stroma, and spread through the blood and lymphatic system to distant sites. Through the process of EMT, cancer cells not only lose their cell–cell adhesions and exhibit elevated motility and invasion, but also gain increased resistance to apoptosis, chemotherapeutic drugs and even develop stem-cell like properties.69 According to current views, EMT has prognostic significance in various cancers as it is highly correlated with invasive phenotype and metastasis-forming capacity. This so-called ‘EMT phenotype’ is a primary determinant of prognosis.70 It has been shown in non-small-cell lung cancer that EMT phenotype is associated with epidermal growth factor receptor mutations and drug resistance,71 and also with formation of cancer stem cells.70 In lung cancer, the circulating tumour cells (CTCs) expressing epithelial marker EpCAM are lower compared with other solid tumours.72 However, when CTC isolation is not based on epithelial markers, the CTC numbers are similar to those of other solid tumours and have strong prognostic value.73 These data suggest that lung cancer CTCs have lost their epithelial characteristics having undergone EMT. A recent review suggests that anticancer and antimetastasis drugs frequently have side effects associated with defective wound healing, implicating that similar molecular pathways contribute to these processes.12 However, despite the growing body of evidence, there is still no direct proof in humans that EMT actually happens as a dynamic process in tumour metastasis formation.
What is the clinical importance of EMT?
Two disease groups need to be considered concerning the clinical importance of EMT in the lung: conditions with fibrosis and tissue remodelling, and malignant lung diseases.
It has been recently highlighted that various forms of lung fibrosis and tissue remodelling are present in all chronic respiratory diseases.74 Current research data highlight that we have to be careful on judging how much EMT contributes to the actual fibrosis, scarring and tissue remodelling. Among the identified targets is the TGF-β pathway and other growth factors, like PDGF, CTGF, FGF and vascular endothelial growth factor, which are considered to trigger EMT and have a proven pathogenic role in fibrotic lung diseases.75–78 Most of these molecular targets have been tested in animal models77 ,79 ,80 but there has been no success so far in translating these findings to the clinic.81 Pirfenidone is the only licensed therapy for IPF82 but the mechanism of action is obscure and there are no in vivo data available of its effects on EMT.
As Wnt signalling pathways have been shown to be important in the pathogenesis in several lung diseases, such as fibrosis,3 asthma,83 COPD84 and lung cancer,85 they could serve as another promising therapeutic target. Currently there are multiple therapeutic approaches targeting Wnt signalling in lung diseases which might be applied in the clinic in the near future.86–88
For a detailed recent review on the involvement of EMT in malignant transformation and cancer progression, see Craene and Berx.70
For a concise summary of recent articles on EMT in lung diseases, please see the table in the online supplement.
Summary and conclusions
In summary, we can say that the extent and significance of EMT in lung diseases is still controversial because of the following issues:
-
A large majority of studies on EMT in lung diseases use methods which take only a ‘snapshot’ of this dynamic process—for example, immunohistochemistry, western blotting or flow cytometry. Although there have been moves to standardise the molecular markers of EMT,5 ,33 there are doubts as to whether EMT can only be defined by the coexpression of certain epithelial and mesenchymal markers66 since similar molecular changes occur in epithelial cells during wound repair.11 ,12
-
In the case of fibrotic changes, genetic lineage tagging of cells in animal models is thought to be a reliable method; however, the results are contradictory in different animal models for lung fibrosis and other organ fibrosis models.33 It seems that the results in transgenic mice differ with regards to the nature of the genetic tag, the method used for its detection and also the quality of the transgenic construct used for recombination.
-
A large proportion of the in vivo data on the connection between fibrosis and EMT have been obtained from the single-dose pulmonary bleomycin challenge model. This has received criticism and clearly there is a need for the development of a more reliable murine lung fibrosis model that accurately reflects human UIP.
-
A growing body of evidence supports that EMT has a pivotal role in malignant transformation, invasion and metastatic capacity, and in the formation of cancer stem cells. In lung carcinomas, CTCs seem to have shifted towards a more mesenchymal phenotype compared with other solid tumours, suggesting EMT. Hitherto, there is no convincing direct in vivo evidence of EMT in human tumours, although suggestions were made about the detection of EMT in tumours in situ.89
In conclusion, much recent research has highlighted the potential of EMT to be involved in lung development and disease responses. The precise clinical importance of EMT outside of tumour biology remains to be determined, but the development of drugs that target growth factors known to promote EMT suggest that modulation of EMT processes in the clinical setting may soon be possible. Whether these are clinically effective remains to be seen.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors DB, NM, RYM, OE and DRT wrote and edited the manuscript.
-
Funding DB is currently funded by the FP7 Marie Curie Intra-European Fellowship (FP7-PEOPLE-IEF 300371) and received funding from the European Respiratory Society Long Term Fellowship (ERS-LTRF 2011–131).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.