Article Text

Abstract
Introduction Evidence suggests that platelets play a significant role in inflammation in addition to their role in thrombosis. Systemic inflammation is linked to poor short and long term outcomes in COPD. Increased platelet activation has been reported in acute exacerbations of COPD (AECOPD). We investigated whether thrombocytosis is independently associated with poor outcomes following AECOPD.
Methods An observational cohort study of patients hospitalised with AECOPD was performed. Patients were >40 years with spirometry confirmed COPD admitted between 2009 and 2011. Platelet count was recorded on admission. The primary outcome was 1-year all-cause mortality. Secondary outcomes included inhospital mortality and cardiovascular events. Analyses were conducted using logistic regression after adjustment for confounding variables.
Results 1343 patients (49% male) were included. Median age was 72 years (IQR 63–79 years). 157 (11.7%) had thrombocytosis. Thrombocytosis was associated with both 1-year mortality and inhospital mortality; OR 1.53 (95% CI 1.03 to 2.29, p=0.030) and OR 2.37 (95% CI 1.29 to 4.34, p=0.005), respectively. Cardiovascular hospitalisation was not significantly increased (OR 1.13 (95% CI 0.73 to 1.76, p=0.600)) in patients with thrombocytosis. Aspirin or clopidogrel treatment correlated with a reduction in 1-year mortality (OR 0.63 (95% CI 0.47 to 0.85, p=0.003)) but not inhospital mortality (OR 0.69 (95% CI 0.41 to 1.11, p=0.124)).
Conclusions After adjustment for confounders thrombocytosis was associated with increased 1-year mortality after exacerbation of COPD. Antiplatelet therapy was associated with significantly lower 1-year mortality and may have a protective role to play in patients with AECOPD.
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is thrombocytosis an independent marker of poor outcome following acute exacerbation of COPD and do antiplatelet therapies improve outcome?
What is the bottom line?
-
Thrombocytosis is independently associated with increased inhospital and 1-year all-cause mortality, and antiplatelet therapy is associated with reduced 1-year all-cause mortality following acute exacerbation of COPD.
Why read on?
-
Thrombocytosis is an accessible, independent predictor of short term and 1-year mortality in acute exacerbations of COPD, and antiplatelet therapy may be associated with a survival benefit.
Introduction
COPD is a systemic disorder associated with increased risk of cardiovascular disease, diabetes and hypertension.1–3 Systemic inflammation is a key driver of cardiovascular disease in COPD.4 ,5 In addition to haemostasis and thrombosis,6 platelets are proposed as key inflammatory mediators.7 ,8 Platelets granules store and secrete a number of bioactive substances which are released upon platelet activation. This results in platelet aggregation, neutrophil and monocyte adhesion, chemotaxis, cell survival and proliferation, coagulation and proteolysis.8
Few studies examine the role of platelets in COPD. In murine models, acute hypoxia has been associated with thrombocytosis.9 Similarly, platelet volume and aggregation are increased in hypoxaemic, stable COPD.10 ,11 Platelet–monocyte aggregates are a sensitive marker of platelet activation, and are increased in patients with COPD, with further increases during acute exacerbations of COPD (AECOPD).12 ,13 With evidence for the pro-inflammatory properties of platelets, and given that COPD is associated with cardiovascular disease,14–16 studies have proposed antiplatelet therapy in COPD.13 ,16 To date, there have been no randomised trials in patients with COPD and there are no studies investigating antiplatelet therapy after AECOPD. We hypothesised that thrombocytosis in patients with AECOPD would be associated with increased short and long term mortality. We also tested the hypothesis that antiplatelets may have a protective role to play in patients with AECOPD.
Methods
Study database
This study describes a secondary analysis of the EXODUS cohort (EXacerbations of Obstructive lung Disease managed in UK Secondary care), a multicentre audit database. The database prospectively included patients admitted to 12 hospitals with AECOPD17 and was approved by the South East Scotland research ethics committee and local Caldicott guardians. Participants were ≥40 years admitted for >12 h from June 2009 to December 2011. Inclusion criteria were: primary diagnosis of AECOPD and a diagnosis of COPD confirmed by spirometry when stable. Exclusion criteria were age <40 years, airways disease primarily due to other cause (community-acquired pneumonia, ILD, asthma, bronchiectasis), suspected or proven alternative diagnosis for the deterioration in symptoms (heart failure, PE or pneumonia on chest X-ray), transfers from other hospitals, and patients in whom active treatment was not considered appropriate (palliative care).
Study data
Demographics and medication records were obtained on admission from patient histories using standard hospital clerk-in pro formas. Laboratory and radiology results included were the first tests from admission. Medications recorded included all COPD and cardiovascular medications. Patients using aspirin or clopidogrel on admission to hospital were recorded as ‘antiplatelet users’. Severity of COPD was classified according to the Medical Research Council (MRC) dyspnoea score, % predicted FEV1 and the Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2011 staging system.18 Missing data were assumed to be normal for the purposes of analysis.
Readmission and postdischarge data were identified from hospital records and morbidity databases using International Classification of Diseases, 10th revision (ICD-10) codes. Data were obtained for 12 months after discharge, and therefore follow-up was completed in December 2012. Patients were entered into the database once, readmissions being recorded as such rather than a new entry.
Laboratory variables including platelet count, white blood cell count and C reactive protein (CRP) were measured according to local protocol with no significant differences in mean platelet counts between hospitals (data not shown). Patients were categorised into three groups for analysis according to platelet count: normal platelet count range; 150–400×109 cells/mm3, thrombocytopenia; platelets <150×109 cells/mm3 and thrombocytosis; >400×109 cells/mm3.
Outcomes
The primary outcome is 1-year mortality, analysed as death from the first day of admission to completion of 1-year follow-up. Secondary outcomes were inhospital mortality and postdischarge mortality (from discharge until 1-year follow-up). Cardiovascular events and cardiovascular mortality were also recorded. Death certification data were used to identify all deaths. Validity of the data was confirmed by manual review of medical records at two sites. ICD 10 codes were used to identify mortality and readmissions due to cardiovascular causes: I20–23 (acute myocardial infarction and acute coronary syndromes including unstable angina), I50 (cardiac failure incorporating congestive cardiac failure (I50.0) and left ventricular failure (I50.1)), I46 (cardiac arrest and cardiac arrhythmia), I44 (atrioventricular disorders), I45 (other conduction disorders), I47 (paroxysmal tachycardia), I48 (atrial fibrillation and flutter) and I49 (other cardiac arrhythmias).
Statistical analysis
Continuous variables were assessed for normality using D'Agostino's K2 test. Categorical variables in tables 1 and 2 were compared between the three platelet groups using the χ2 test. As continuous data in tables 1 and 2 were found to be non-parametric, these data were compared using the Kruskal–Wallis test. Correlations of platelet count with CRP and white blood cell count were performed using simple linear regression. Cumulative survival was determined by the Kaplan–Meier method and compared using the log-rank test.
Demographic data
Exacerbation characteristics
Logistic regression analysis
Logistic regression was used to generate ORs for the association between thrombocytosis or antiplatelet therapy with mortality. For the primary logistic regression analysis, covariates were selected based on clinical assessment of the likelihood of confounding and retained in the final model if they remained significant at p<0.05 or were found to modify the OR by at least 10% when included in a model with the main exposure variable. Selected variables for the analysis of thrombocytosis were age (continuous), gender (male=1/female=0), residence in long term care (yes/no), history of myocardial infarction (yes/no), stable angina (yes/no), coronary artery bypass grafting (yes/no), stroke (yes/no), congestive cardiac failure (yes/no), neoplastic disease (yes/no), diabetes (yes/no), current use of antiplatelet drugs (yes/no), Body Mass Index (continuous), FEV1% predicted (continuous), MRC dyspnoea score (continuous), use of long term oxygen therapy (LTOT) (yes/no), current cigarette smoking (yes/no), PaCO2 (continuous), acidosis (yes/no), PaO2 to FiO2 ratio (continuous) and severity of exacerbation using the BAP-65 (blood urea nitrogen >25 mg/dL, altered mental status, pulse >109 beats/min, age >65 years) severity index, a validated COPD exacerbation index.19 In addition, for the analysis of antiplatelet therapies effect, modification of use of β-blockers (yes/no), statins (yes/no), ACE inhibitors (yes/no) and anticoagulants (yes/no) was tested. The Hosner–Lemeshow goodness of fit test was used to assess model adequacy. The data are reported in accordance with STROBE guidance.
A p<0.05 was considered statistically significant for all analyses. All data were analysed using SPSS V.21 for windows.
Results
A total of 1698 unique records were entered into EXODUS; 355 were excluded, primarily due to inclusion more than once (n=131) or loss to follow-up where data could not be verified with long term follow-up data (figure 1).

Flow chart describing the study. AECOPD= acute exacerbations of COPD. * Patient data were rejected if they contained clear inaccuracies that could not be resolved with the researchers or lacked an accurate identification code to enable record linkage for determining outcomes (therefore classified as lost to follow-up).
Overall, 1343 patients (49% male) were included in the study. Demographics, COPD severity, comorbidities and medication use are shown (table 1). Of the cohort, 85.4% of patients had a normal platelet count (median platelets 243×109 cells/mm3, IQR 199–299×109 cells/mm3), 2.9% were thrombocytopenic (117, IQR 97–141×109 cells/mm3) and 11.7% had thrombocytosis (474, IQR 433–547×109 cells/mm3) on admission. As the thrombocytopenia group was small, no further analysis was performed but data are shown (tables 1 and 2). There was no difference in GOLD class between the groups (table 1). Other recognised markers of severity were more common in the thrombocytosis group compared with the normal group, with higher frequency of exacerbations in the previous year (mean 1.42 SD 1.9 vs mean 1.1 SD 1.8, p=0.0240), a lower median albumin (34 g/L IQR 31–38 vs 37 g/L 33–40, p<0.0001) and lower Body Mass Index (table 1). When comparing thrombocytosis and normal platelet groups (excluding thrombocytopenia), thrombocytosis was significantly associated with male gender (p=0.02), use of LTOT (p=0.03), higher white blood cell count (p<0.0001) (table 2), more frequent acidosis (p=0.01), higher PaCO2 values (p=0.008), lower PaO2 to FiO2 ratio (p=0.0003) and more use of non-invasive ventilation (p=0.01) (table 3).
Thrombocytosis and outcomes: logistic regression analyses
Length of stay was similar between patients with thrombocytosis (median 5 days, IQR 3–8) and patients with normal platelet count (median 6 days, IQR 3–11) and thrombocytopenia (7 days, IQR 2–11), p=0.165.
The impact of thrombocytosis on mortality and cardiovascular events
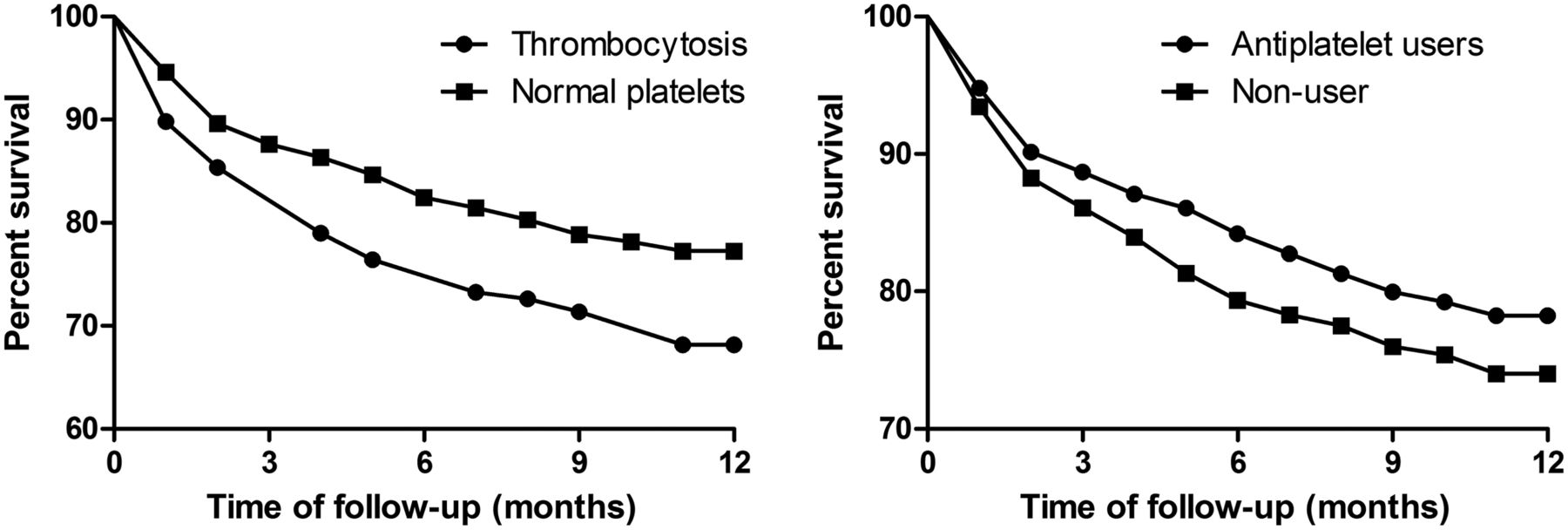
Inpatient mortality was 6.2% and 18.8% of patients who survived beyond hospital discharge had died at 1 year. Thrombocytosis was associated with increased mortality over 1-year follow-up (OR 1.53 (95% CI 1.03 to 2.29, p=0.030)). The logistic regression model and significant covariates predicting 1-year mortality are shown (table 3). These were age, congestive cardiac failure, neoplastic disease, MRC dyspnoea score, LTOT and acidosis during exacerbation, all of which were associated with increased mortality, while antiplatelet therapy was associated with reduced mortality (analysed in more detail below). The Kaplan–Meier survival analysis is shown in figure 2. The difference between the thrombocytosis and normal platelet group was statistically significant by the log-rank test (p=0.008).

{kind=link}
{kind=link}
Kaplan–Meier survival curve comparing the thrombocytosis and normal platelet groups. Curve comparison by log-rank test, p=0.009 for thrombocytosis versus normal platelet counts, p=0.09 for antiplatelet users versus non-users.
Thrombocytosis patients had increased inhospital mortality compared with those with normal platelet counts (11.5% vs 4.9%). In the logistic regression analysis, thrombocytosis was independently associated with increased inhospital mortality (OR 2.37 (95% CI 1.29 to 4.34, p=0.005)). When considering only events after hospital discharge to 1 year, the OR for mortality for platelet count >400 was 1.30 (95% CI 0.82 to 2.06, p=0.261).
Overall, 268 patients (20%) were hospitalised due to a cardiovascular event during follow-up. These were predominantly hospitalisation associated with congestive cardiac failure (11%), acute coronary syndromes (5%), acute myocardial infarction (4%) and/or arrhythmias (17%). Increased 1-year mortality in patients with thrombocytosis could not be attributed solely to cardiovascular events because there was no statistically significant increase in cardiovascular hospitalisation (OR 1.13 (95% CI 0.73 to 1.76), p=0.6). The strongest predictors of cardiovascular hospitalisation in this model were age (OR 1.02 (95% CI 1.01 to 1.04, p=0.006)), previous myocardial infarction (OR 2.23 (95% CI 1.55 to 3.21, p<0.0001)), neoplastic disease (OR 2.00 (95% CI 1.33 to 3.00, p=0.0009)), diabetes (OR 1.79 (95% CI 1.15 to 2.80, p=0.011)), MRC dyspnoea score (OR 1.69 (95% CI 1.44 to 1.98, p<0.0001)) and LTOT (OR 2.35 (95% CI 1.54 to 3.59, p<0.0001)).
Antiplatelets and anticoagulants
To further investigate the role of platelet function in AECOPD, we studied whether antiplatelet therapy would influence outcomes. A total of 713 (53.0%) patients were using antiplatelet agents. In the Kaplan–Meier analysis, there was no significant relationship between antiplatelet therapy and mortality (p=0.093 by log-rank test, figure 2). After adjustment for confounders in the logistic regression analysis (shown in table 4), antiplatelet therapy was associated with reduced overall 1-year mortality after exacerbation of COPD (OR 0.63 (95% CI 0.47 to 0.85, p=0.003)) but was not significantly associated with inhospital mortality (OR 0.69 (95% CI 0.41 to 1.11, p=0.124)). Mortality was significantly increased in patients who survived to hospital discharge up to 1 year (OR 0.62 (95% CI 0.46 to 0.85, p=0.003)). The relationship between antiplatelet therapy and cardiovascular hospitalisation (OR 0.73 (95% CI 0.50 to 1.06, p=0.097)) and cardiovascular death (OR 0.77 (0.47–1.28, p=0.311)) was not statistically significant. To explore whether the apparent benefit observed was an antiplatelet or anticoagulant effect, warfarin was also assessed (n=98) with no significant difference in 1-year overall mortality (OR 0.96 (95% CI 0.56 to 1.63, p=0.877)).
Antiplatelet therapy and outcomes: logistic regression analyses
Systemic inflammatory markers and outcomes
There was a weak correlation between white cell and platelet counts (R=0.158, p<0.0001). CRP showed no significant correlation with platelet count (p=0.5). After adjustment for confounders in a logistic regression model, CRP was not associated with either total 1-year mortality (OR 1.01 95% CI 0.98 to 1.04 per 10 mg/L increment, p=0.455) or inhospital mortality (OR 1.03 95% CI 0.99 to 1.08, per 10 mg/L increment, p=0.107). There was no relationship between CRP and cardiovascular events (OR 1.01 (0.97–1.03, p=0.912)). This was not affected by entering CRP as a categorical variable using prespecified cut-offs (>50 mg/L or 100 mg/L, data not shown).
Similarly, white blood cell count was not an independent predictor of either total 1-year mortality (OR 0.98 (95% CI 0.95 to 1.01), p=0.222) or inhospital mortality (OR 0.95 (95% CI 0.89 to 1.01), p=0.087). There was no relationship between WCC and cardiovascular events (OR 1.00 (95% CI 0.98 to 1.02), p=0.854). Entering white blood cell count as a categorical variable using prespecified cut-offs (>12×109 cells/mm3 or >15×109 cells/mm3) also did not identify a statistically significant relationship with any of the outcomes (data not shown).
Discussion
Our data demonstrate thrombocytosis is associated with significantly increased 1-year mortality following admission with AECOPD. Thrombocytosis was associated with significantly increased inhospital mortality and correlated with markers of type II respiratory failure and exacerbation severity. Thrombocytosis was not associated with chronic comorbidities such as ischaemic heart disease, diabetes or renal impairment.
As with all association studies, our data cannot prove causation and the underlying mechanisms between thrombocytosis and death are not clear. McDonald et al demonstrated that 1–4 days of hypoxia resulted in thrombocytosis, and hypothesised that ‘shedding’ of immature platelets from megakaryocytes was the mechanism.9 This hypothesis is supported by Wedzicha et al who established a negative correlation between mean platelet volume (MPV) and arterial oxygen tension,10 given that immature reticulated platelets have a larger MPV than mature circulating platelets. Larger, immature platelets have also been shown to be more haemostatically active than mature platelets,20 which may also explain the increased platelet aggregation found in hypoxaemic COPD patients,11 and increased platelet activation in patients with AECOPD.12 In our cohort, respiratory failure is also indicated as an important factor in the association between thrombocytosis and increased mortality. Baseline characteristics were very similar between groups although there was a small difference in the frequency of LTOT prescription comparing the thrombocytosis and normal platelet groups. Similarly, the most striking differences between the nature of acute exacerbation pertain to acute ventilatory failure with thrombocytosis patients more likely to be hypoxic, hypercapnic or receive non-invasive ventilation. While acute respiratory acidosis is a strong predictor of mortality in AECOPD,21 the effect of thrombocytosis was independent of this, suggesting that patient specific haematological responses to hypoxia and respiratory failure may be more important than previously recognised. This individualised response to infection or another exacerbation trigger may mark out patients more likely to have a pro-inflammatory response to subsequent exacerbations and explain the observed increase in all-cause mortality.
Given the known association between ischaemic heart disease and COPD,22–24 we expected thrombocytosis to be strongly associated with cardiac comorbidity and mortality. Hypoxic pulmonary vasoconstriction with increase in cardiac afterload, strain on the right ventricle and sequential tachycardia has been postulated as an additional mechanism for arrhythmia and cardiovascular events.24 Despite this, we did not demonstrate a significant increase in cardiac hospitalisations and death. It is therefore difficult to propose cardiac disease as the key mechanism to explain excess morbidity and mortality seen with thrombocytosis and supports a mechanism more closely related to hypoxia and inflammation. In our study, CRP, another common inflammatory marker, was not correlated with an increase in mortality. Similarly, previous studies exploring CRP as a possible marker of severity and mortality in AECOPD have been equivocal.25 ,26 It may be that platelet responses and CRP represent different aspects of the inflammatory response. The finding that acute changes in platelet count during AECOPD are associated with increased 1-year all-cause mortality suggests that the process is more complex than an acute inflammatory insult. The ECLIPSE cohort (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints) demonstrated the complexity of chronic inflammation in COPD, and brought home the concept of persistent systemic inflammation during periods of stability.27 It may be that acute haematological responses are more important to long term inflammation than more commonly recognised acute phase proteins such as CRP.
In the whole cohort, patients on antiplatelets had a significant reduction in 1-year mortality that again was not solely attributable to a reduction in cardiovascular events. In observational studies, there is always potential that a signal is due to the ‘healthy user effect’, even when analyses adjusted for comorbidities. We regard this explanation as unlikely since antiplatelet therapies are generally prescribed for cardiovascular and cerebrovascular diseases which are associated with increased mortality, and therefore the effect of antiplatelets may be potentially underestimated.28 When combined with recent data highlighting the importance of other cardiovascular treatments in COPD, and particularly β-blockers,29 ,30 there is a growing argument that physicians should be seeking and treating cardiovascular risk in COPD. Large scale randomised control trials in antiplatelets are warranted as they may prove a role for inexpensive therapies with considerable benefits in COPD.
To our knowledge, this is the first prospective study to examine thrombocytosis in AECOPD. EXODUS is a large, multi-centre database with similar demographics to that of the 2008 national BTS audit31 and we therefore feel that the results should be generalisable to other populations. Nonetheless, we must acknowledge several limitations. It is an observation and therefore prone to the inherent weaknesses of this type of study. ICD-10 codes were used and are known to have limitations in accuracy. In particular, we used death certification to classify mortality as cardiovascular or non-cardiovascular which may have modest accuracy.32 However, this method has been well used in similar studies and any inaccuracy is likely to be similar in both the thrombocytosis and normal platelet group. Another potential weakness is that data were only recorded from the day of admission. It would have been desirable to have serial platelet counts to assess whether thrombocytosis was transient, related only to that admission or whether there were persistent or recurrent abnormalities in platelet counts. The association of thrombocytosis with outcome was independent of neoplastic disease suggesting that this was an effect of exacerbations of COPD, rather than due to neoplastic disease or haematological malignancy. Thrombocytosis was significantly associated with outcome even after exclusion of patients with very high platelet counts further strengthening this conclusion. Although it would be useful to have included more detailed assessment of platelet and clotting function rather than the crude numbers, platelet count is readily available to all clinicians on a day-to-day basis. We also did not follow-up medication use, and therefore cannot guarantee that all patients who were on antiplatelets on admission were still prescribed them throughout the study. The reasons for discontinuation of antiplatelet agents are few, however, and we hypothesise that this may only have been a very small number of patients and unlikely to have affected our results. Finally, while the observation of apparent risk reduction associated with platelet therapy is of great interest, randomised controlled trials would be required to confirm or refute its usefulness in patients suffering from AECOPD.
Conclusions
In AECOPD, thrombocytosis is associated with increased mortality. Acute type II respiratory failure and LTOT were more common in patients with thrombocytosis, though other severity markers were similar. These results suggest platelets may have an important inflammatory role in COPD, possibly related to hypoxia. In all patients, antiplatelet medications were associated with significantly lower 1-year mortality and may have a protective role to play in patients with AECOPD.
Acknowledgments
We thank Ashan R Akram, University of Edinburgh; Ross Archibald, University of Dundee; Louise Peet, University of Dundee; Charly Sengheiser, Doncaster Royal Infirmary; Duneesha De Fonseka, Doncaster Royal Infirmary; Gillian B Fleming, Edinburgh Royal Infirmary; Hazel Rooney, Perth Royal Infirmary; Duncan Mills, Royal Infirmary of Edinburgh; Sarah Higgins, Doncaster Royal Infirmary; John Corcoran, John Radcliffe Hospital, Oxford; Joseph Hodgson, NHS Tayside; Martin K Smith, NHS Borders; and Mudher Al-Khairalla, Doncaster Royal Infirmary, for their help with data collection.
References
Footnotes
-
Contributors MTH was responsible for study conception, design, data collection and data analysis. PS, PW and AS were responsible for data collection and data analysis. JDC was involved in the study design, data collection and data analysis. SS was involved in study design and data analysis. All authors contributed to drafting the manuscript and approved manuscript prior to submission. All authors had full access to all of the data in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. JDC is the study guarantor.
-
Competing interests None.
-
Ethics approval South East Scotland research ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.