Article Text

Abstract
Lung function tracks from the earliest age that it can be reliably measured. Genome wide association studies suggest that most variants identified for common complex traits are regulatory in function and active during fetal development. Fetal programming of gene expression during development is critical to the formation of a normal lung. An understanding of how fetal developmental genes related to diseases of the lungs and airways is a critical area for research. This review article considers the developmental origins hypothesis, the stages of normal lung development and a variety of environmental exposures that might influence the developmental process: in utero cigarette smoke exposure, vitamin D and folate. We conclude with some information on developmental genes and asthma.
- Asthma
- Asthma Genetics
Statistics from Altmetric.com
Introduction
In this review we consider several areas where environmental exposures, genomics, development and asthma occurrence overlap. We will first consider the developmental origins of disease hypothesis and then we cover the anatomic stages of normal fetal lung development and resources developed to understand the genomic origins of asthma. We then consider the transcriptomic profile of human lung development, in utero cigarette smoke exposure and its effects on fetal lung development and asthma, expression quantitative trait loci (eQTL) identification, the effects of folate and vitamin D on fetal lung development and asthma, and finish with some conclusions and next steps.
Developmental origins of health and disease hypothesis
The developmental origins of health and disease (DOHaD) hypothesis suggests that exposures during early life from conception into early childhood can have an influence on an individual's health and disease risk in adult life.1 Although the initial hypothesis as proposed by Barker related coronary artery disease to fetal under-nutrition, it has now been applied to a variety of diseases, including the development of childhood asthma. The first evidence of in utero factors influencing long-term disease risk came from a large study demonstrating the association between an increased risk of coronary artery disease with low birth weight.2 Three factors were believed to explain the higher disease incidence in low-birth-weight babies: fewer cells in key organs, a ‘thrifty’ way of handling food, and a higher vulnerability.3 These findings are thought to be a result of developmental plasticity, which is defined as the ability of one genotype to give rise to a number of different phenotypes in response to environmental exposures that occur during development.3 ,4 It is now known that a number of additional environmental factors can play a role in the in utero programming of fetal development, including lung development, which subsequently increases disease susceptibility in childhood and adulthood. Since gene expression changes that result from intrauterine environmental exposures and epigenetics are a potential explanation for the DOHaD hypothesis,5 genomic technologies are now being applied in the context of development to understand the mechanisms underlying the fetal origins of disease. In this review we examine the existing evidence for the genomics of lung development and the fetal origins of asthma.
Human fetal lung development
Human lung development starts at approximately the fourth week of gestation and continues into the postnatal period. The developing human fetal lung can be classified into six temporally overlapping successive stages based on gross morphological changes and histological appearance: embryonic (4–7 weeks), pseudoglandular (7–17 weeks), canalicular (17–26 weeks), saccular (27–36 weeks) and alveolar (36 weeks–2 years) (figure 1).6 Although the exact timing of the transitions between histological stages is variable in the existing literature, the sequence of the stages is universally agreed upon. Branching morphogenesis, which is the genetically programmed process of airway development, occurs during the pseudoglandular and canalicular stages of development. Unlike in the mouse in which alveolar development occurs exclusively in the postnatal period, alveolar development in the human commences at approximately 36 weeks post conception and is only complete in adolescence. Airway growth also continues throughout the postnatal period, and into adolescence.

Stages of human lung development. This figure from Kajekar76 depicts the known histological stages of human fetal lung development. Republished with permission from Elsevier.
Resources developed to understand the genomic origins of asthma
High throughput genomic technologies are being used with increasing frequency in the study of complex diseases. The use of these resources in the context of development biology, however, is still in its infancy. Resources that can help us better understand the changes that alter the course of lung development include tissue-specific gene expression, genotype, epigenetics, proteomics, metabolomics and other data spanning the developmental timeline. By applying high throughput genomic methodologies including genotype, gene expression and DNA methylation to fetal lung and placental tissue that was obtained through a collaborative research effort, we hope to further elucidate the genomic trajectories of human lung development. To add to this effort is the availability of genotype and gene expression data from subjects with asthma in large population cohorts such as the Childhood Asthma Management Program (CAMP). The NCBI Gene Expression Omnibus also has a large number of asthma-related datasets which have been obtained from tissues such as bronchial biopsies, CD4 T cells, CD8 T cells, peripheral blood mononuclear cells and cultured airway smooth muscle cells, and can allow researchers to perform integrative bioinformatics analyses. Murine data further add to our understanding of development and disease, allowing researchers to fill in missing developmental time points and to verify genomic targets. Murine models of asthma and in utero exposures have been useful in understanding mechanisms and have allowed time series analyses, which are not possible in human subjects.
The transcriptomic profile of human lung development
Human fetal lung development results from a complex interacting system of genetic, mechanical and molecular processes, which are unlikely to be captured by the traditional histological stage alone. Furthermore, the biological mechanisms underlying lung development have not been completely characterised.7–9 Several studies have investigated the gene expression profile of the developing mouse lung.10–13 These models have begun to define the complex regulation of gene expression that occurs during lung development and have provided insight into the molecular determinants of normal lung development. In spite of the advances made in our understanding of lung development that have resulted from murine models of development, there remains a paucity of data regarding the gene expression profile of lung development in the human. Using genome-wide gene expression profiles from 38 human fetal lung tissue samples from postconception ages 53–154 days, we have previously identified global patterns of gene expression in human lung development.14 Interestingly, principal components analysis (PCA) of human fetal lung tissue gene expression identified a molecular transition phase that was independent of histological stage,14 demonstrating the promise of the application of genomic technologies to the study of development to elucidate novel biology (figure 2).

Novel developmental growth stage identified by genomics. In this figure from Kho et al13 the transcriptome profile of the 38-sample developing human fetal lung time series is shown in the first two principal components. While age correlates well with PC1, inflection points in PC2 indicate a novel molecular transition at day 94, which is independent of histological stage. Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Kho et al (2009) Expression profiles of the mouse lung identify a molecular signature of time-to-birth. Am J Respir Cell Mol Bio 40:47–57. Official Journal of the American Thoracic Society.
Supervised data analysis strategies explicitly use a priori knowledge or assumptions about the system in addition to the given (expression) data. For example, determining genes that are differentially expressed between pseudoglandular and canalicular stages requires the respective time intervals for these stages as computational input variables/parameters; identifying genes that have a specific expression profile requires a hypothetical profile template against which all genes are compared (with no guarantee that a gene exists in the system possessing that hypothetical profile); and investigating the expression profile of genes involved in branching morphogenesis requires knowledge of gene attributes from biological knowledge repositories such as PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), the Gene Ontology Consortium (http://www.geneontology.org/) or KEGG (http://www.genome.jp/kegg/).
In contrast, unsupervised data analysis strategies use the given data as their sole input variables/parameters. These data are typically represented as an algebraic matrix of N genes by M sample time points with real number entries representing the measured expression of a specific gene for a particular sample. For example, identifying the direction and magnitude of variance between the M samples in N-dimensional gene (transcriptome) space is a mathematical operation purely involving N subjects in M object feature space, with no knowledge of individual sample or gene biology, which is the case when one employs linear algebraic singular value decomposition-based methods such as PCA.15 ,16 We have used PCA to identify the dominant transcriptome-scale sample variations between developing lung time points and found that these variations (called principal components) correlate with morphological/physiological events in the developing lung such as developmental age, time to birth and the transition between the classical developmental stages.13 ,14 Interestingly, this more agnostic analysis strategy revealed that aside from a large-scale transcriptomic phase transition around days 110–117 post conception that coincides with the transition between the pseudoglandular and canalicular stages, the pseudoglandular stage time interval contained an earlier large-scale transcriptomic phase transition at around days 91–96 post conception. This phase transition corresponded to a sharp increase in the expression of genes with immunological major histocompatibility complex class II and surfactant function ontologies. In addition, this approach helped to define what we call a developing lung characteristic gene set (abbreviated DLCGS) using genes that were the top 5% contributors in the three most dominant principal components. The DLCGS from different developing lung transcriptome datasets were found to be significantly enriched for known ontologies related to lung morphogenesis, structure and function, further validating this approach.
One obvious way to investigate a genetic connection between development and (chronic, non-cancer) disease is to perturb a candidate disease gene in a cognate developmental model (eg, cell culture or animal) and to look for any associated phenotypes. Vice versa, one can investigate a known development-critical gene for any perturbations in a relevant disease-control subject population. Besides being single molecule or gene-centric, the limitations of cell culture or animal models of human disease are well known.17 Candidate disease genes and genetic loci can be obtained from a variety of sources but one recent development is the use of genome-wide association studies (GWAS) for this purpose.
Association of lung development genes with asthma and lung function in populations with asthma
The application of genomic technologies to development has allowed us to start to identify the biological mechanisms underlying the fetal origins of complex diseases like asthma. Using genome-wide gene expression profiles from human fetal lung tissue samples from the pseudoglandular and canalicular stages of human lung development, we identified genes in the Wnt-signalling pathway that were differentially expressed by histological stage and that harbour genetic variants that are associated with impaired lung function in two childhood asthma cohorts.18 In addition, using genome-wide gene expression profiles of human and mouse lung development, we have also demonstrated differential expression of several asthma genes previously identified in GWAS of asthma, including ROBO1, RORA, HLA-DQB1, IL2RB and PDE10A.19 Furthermore, there was a trend towards over-representation of known asthma genes among genes differentially expressed during human and mouse lung development.19 These results suggest that gene expression profiling during lung development can be used to identify genes implicated in the pathogenesis of respiratory diseases like asthma and may be used to elucidate the molecular underpinnings of this complex respiratory disease. In addition, this approach demonstrates the pleiotropy of human genes in that genes involved in critical developmental steps are also involved in disease pathogenesis.
Alternatively, one can apply a less gene-centric, more meta-analytic and bioinformatics based approach by investigating overlaps between sets of genes (or genetic loci) implicated in specific disease and developmental contexts to infer development–disease connections and prioritise genes for further functional validation. We previously studied overlaps between the set of all asthma susceptibility genes reported in PubMed relative to different developing lung time series transcriptome data (DLCGS).19 While the overlaps were not all significant, the ORs were consistently >1.0, and a common set of genes showed up in asthma–development set intersections.
The role of in utero smoke in fetal lung development genomics and the development of asthma
There is substantial evidence to suggest that in utero smoke (IUS) exposure results in chronic respiratory disease in the postnatal period. IUS exposure has been associated with reduced lung function and lower respiratory system compliance at birth.20–22 Interestingly, airflow obstruction that results from IUS exposure persists into early childhood and beyond.23–26 Furthermore, IUS has been associated with the risk of recurrent wheeze27 and incident asthma in school age children26 ,28 ,29 and has been associated with asthma during adulthood.30 ,31 In addition, in children with asthma IUS exposure results in decreased lung function32 and a reduced response to inhaled corticosteroids on airway responsiveness.33 Several proposed mechanisms that explain the association of IUS with asthma susceptibility include direct injury to the fetus that results from IUS exposure, alterations of gene expression or gene-by-environment interactions that result from IUS exposure. However, the biological mechanisms underlying the association of IUS and asthma susceptibility are unclear.
Animal studies demonstrate that IUS results in structural effects on the developing lung34 and decreased fetal lung growth.35 Data from non-human primate studies of IUS suggest that IUS exposure increases airway wall dimensions and alterations in lung mechanics.36 In humans, fetuses exposed to maternal smoking during pregnancy have notable hyperplasia of the pulmonary neuroendocrine cells.37
To date, the vast majority of what we know about the effect of IUS on gene expression is a result of animal models. Data from non-human primate studies suggest that IUS exposure increases collagen type I and III deposition in airway and alveolar walls.36 Mouse models of IUS exposure demonstrate the nicotine exposure during early development leads to increased surfactant proteins A and C expression, resulting in increased branching morphogenesis.38 Mouse models of IUS exposure have also demonstrated significant differential expression of 80 transcripts in the lung of adult mice exposed to IUS, including genes involved in immune response, cell proliferation and apoptosis.39 Furthermore, several of the genes that were differentially expressed in the lung of mice exposed to IUS included genes that have been recognised to induce lung inflammation, including arginase 1 (Arg1) and genes in the chitinase family.39
In human subjects, the genomic effects of IUS is less certain. Children who were exposed to IUS and were homozygous for the Arg16 allele of the β2-adrenergic receptor had a threefold increased risk of lifetime wheeze compared with non-exposed subjects.40 These results suggest that environmental exposures including IUS must be considered when evaluating the effect of certain genotypes on respiratory health outcomes.40
To assess the effects of IUS on human fetal lung development, we along with our collaborators determined that placental cotinine, a nicotine metabolite, can be used to classify fetal tissues by their IUS exposure status.41 We identified that an optimal cut point of 7.5 ng/g of placental cotinine had a sensitivity of 78.7% and specificity of 100% to classify placenta samples from mothers who smoked versus those from mothers who did not smoke (figure 3).41 Using placental cotinine values as a surrogate for IUS exposure, we next demonstrated that the expression of xenobiotic-metabolising enzymes (CYP1A1 and CYP1B1) were significantly induced by IUS in human fetal lung tissue.41 Using genome-wide gene expression profiles on human fetal lung tissue samples (post-conception age 53–137 days) with measured placental cotinine values as a biomarker of IUS exposure, we performed a differential gene expression analysis between IUS exposed (n=104) and unexposed (n=104) fetal lung tissue. We identified 154 genes that were differentially expressed between IUS exposed and unexposed subjects (adjusted p<0.05). Interestingly, several microRNAs showed the highest correlation with the IUS exposure (p=2×10−4) (unpublished data). These results suggest that altered gene expression that results from IUS exposure during lung development may be the biological basis for the association of IUS with asthma, however further investigation of these data is currently underway.

Cotinine distribution in human fetal lung tissue. This histogram from Vylidahl and colleagues41 demonstrates the distribution of cotinine, a known nicotine metabolite, in human fetal lung tissue samples. Republished with permission from American Society for Pharmacology and Experimental Therapeutics.
eQTL mapping in development
eQTL mapping seeks to identify genetic polymorphisms that influence gene expression levels and has been used in a variety of disease contexts. Recently, Francesconi and Lehner performed eQTL mapping in Caenorhabditis elegans to identify how genetic polymorphisms influence dynamic gene expression patterns that occur during development.42 They demonstrated that by incorporating dynamic variation they were able to identify >50% more eQTLs, and that genetic polymorphisms extensively affect the timing, rate, magnitude and shape of gene expression changes during development.42 This analytic approach has yet to be performed in lung development and warrants further evaluation in this context.
Using an integrative genomic approach in human fetal lung development, we are starting to identify genetic variants that influence in utero gene expression that subsequently contributes to asthma susceptibility. We screened for cis-acting single nucleotide polymorphism (cis-eQTL) by testing variants mapping within 50 kb of expressed transcripts for association with transcript abundance using generalised linear models adjusted using principal components. We identified genetic variation influencing in utero gene expression of 592 genes in the human fetal lung. Of the significant cis-eQTLs identified, 39 were associated with lung function in subjects with asthma participating in the CAMP (unpublished data). The T allele of the most significant cis-eQTL (rs2392246) was associated with increased BBS9 expression and a lower forced expiratory volume in 1 s (FEV1) (p=0.0006) in subjects with asthma. Interestingly, BBS9 is a protein complex required for ciliogenesis. Although preliminary in nature, our results suggest that mapping of regulatory genetic variation in human fetal lung tissue is a powerful method for identifying genes that influence impaired lung function in subjects with asthma. Integrative genetic approaches in human fetal lung development can help identify such variation and enhance our understanding of the genetic basis of lung function impairment. Furthermore, these results provide further evidence for the fetal origin of impaired lung function.
The role of folic acid in the development of asthma
Folate is part of the water-soluble B complex family of vitamins and includes the natural form and the fully oxidised monoglutamate form used in dietary supplements.43 Folate plays a role as a methyl donor for S-adenosylmethionine44 and may thus influence DNA methylation. Methylation is also required for the synthesis of thymidine monophosphate from deoxyuridine monophosphate, thereby preventing the incorporation of uracil in DNA and preventing point mutations, single and double stranded DNA breaks, chromosome breakage and micronucleus formation.45 Peri-conceptional folic acid supplementation has been shown to prevent neural tube defects46 ,47 and the US Public Health Service recommends that all pregnant women should receive 400 μg of folic acid daily. Studies assessing the relation of maternal folate intake and DNA methylation in humans have yielded conflicting results.48 ,49 Recently there has been a growing interest in the relationship between maternal folate supplementation and the development of asthma in the offspring based on animal studies, whereby supplementation with methyl donors during pregnancy was associated with the development and severity of reactive airway disease in the offspring.50 However, as concluded by a recent review and meta-analyses there is not enough evidence to support the association of peri-conceptional folate intake and development of asthma in the offspring at this time.51
The role of vitamin D in fetal lung development genomics
The role of vitamin D in fetal lung development and its association with asthma has been investigated clinically and through cellular and animal models. The combined evidence suggests that vitamin D plays an important role in utero that is related to future risk of asthma.
The association of low maternal vitamin D on later development of childhood wheezing and asthma has been examined in multiple birth cohort epidemiological studies. In a Scottish birth cohort of 1212 individuals, there was an increased risk for childhood wheezing at 5 years that was inversely associated with maternal vitamin D intake during pregnancy.52 Camargo et al53 followed a Massachusetts pre-birth cohort of 1194 children in Project Viva and found that higher maternal vitamin D intake during pregnancy was associated with a lower rate of recurrent wheeze in children at 3 years. In a Japanese cohort of 763 mother–child pairs, a threshold effect was found at which children of mothers who consumed 4.309 mg or more of vitamin D per day during pregnancy had a significantly reduced risk of wheeze at 16–24 month follow-up.54 More recently a prospective study in 44 825 Danish mother–child pairs in the longitudinal Danish National Birth Cohort examined vitamin D intake in mid-pregnancy, finding a weak inverse relationship between high total vitamin D and asthma at 7 years, without data to suggest a threshold of vitamin D intake that was associated with reduced risk.55
Cord blood was examined as part of a prospective birth cohort of 922 from New Zealand, revealing that low levels of cord-blood 25-hydroxyvitamin D were associated with an increased risk of childhood wheezing at 5 years, but not the incidence of asthma.56 Conversely, a birth cohort of 178 followed in the UK reportedly demonstrated that elevated maternal blood 25-hydroxyvitamin D in the third trimester was associated with an increased likelihood of asthma at 9 years, though notably the response rate was only 30% and confounders were not fully adjusted for.57 ,58
A systematic review and pooled meta-analysis examined many of these studies and found that higher maternal vitamin D intake was associated with reduced odds of wheezing (OR 0.56, 95% CI 0.42 to 0.73; p<0.001). This pooled analysis did not show an association with increased risk of asthma.59
Newer studies have tested these results. Goldring et al60 performed a randomised trial following the offspring of 158 pregnant women in the UK. They found that vitamin D supplementation in the third trimester had a modest effect on cord blood vitamin D levels but no association with decreased wheezing or lung function measured by impulse oscillometry in offspring at 3 years of age.60 An ongoing randomised trial of vitamin D supplementation during pregnancy will provide further insight into the relationship between maternal vitamin D intake and development of asthma in children (Randomised trial: maternal vitamin D supplementation to prevent childhood asthma (VDAART), ClinicalTrials.gov: NCT00920621).
Fetal and animal studies have further explored the mechanism behind this clinical association. There is evidence that vitamin D plays an important role in the regulation of fetal lung growth and development.61–63 Gaultier et al64 showed that rachitic rat pups born to vitamin D deficient mothers had normal lung weights but decreased lung compliance consistent with abnormal alveolar development. Edelson et al63 demonstrated that vitamin D3 is a growth factor for alveolar type II cells and suggested that vitamin D may be involved in local control of epithelial proliferation related to lung development.
Receptors for 1,25-dihydroxy vitamin D have been characterised in fetal alveolar epithelial cells, and it has been shown that fetal lungs decrease glycogen content and increase surfactant synthesis and secretion after exposure to 1,25-dihydroxy vitamin D.65–67 Vitamin D acts as a mediator of epithelial–mesenchymal cell interactions. In fetal rat lung there is a paracrine system that is activated during the last 3 days of pregnancy, promoting lung fibroblasts to produce 1,25-dihydroxy vitamin D3. Type II pneumocytes have vitamin D receptors that respond to 1,25-(OH)2D3 by increasing surfactant synthesis/secretion and reducing glycogen content.61 ,62 ,68 Sakurai et al69 investigated the important role vitamin D plays in modulating lipofibroblasts during lung development. They proposed a model for the alveolar vitamin D paracrine system and suggested a mechanism for the increased surfactant synthesis and resultant alveolar septal thinning during perinatal lung maturation.
It has been demonstrated that poor lung development is a risk factor for asthma in childhood.70 Zosky et al71 showed a mouse model of in utero vitamin D deficiency in which offspring from vitamin D-deficient mothers had impaired lung volume and corresponding deficits in lung function that could not be explained by the decreased lung volume alone. This provides evidence for a potential mechanism for the association between gestational vitamin D deficiency and the development of future obstructive lung disease.71 Follow-up work by this group demonstrated that in utero vitamin D deficiency suppressed the proliferation of airway-draining lymph node cells and their ability to secrete T-helper cell type 2 cytokines such as IL-3 and IL-5 in male and female mice. Additionally, male offspring with vitamin D deficiency from the point of conception had enhanced numbers of eosinophils and neutrophils on bronchoalveolar lavage, an effect that was reversible with postnatal supplementation. In this study it was not possible to distinguish the effects of maternal vitamin D status versus the role of the vitamin D status of the individual after birth on lung development.72
A recent publication by our group further explored the mechanisms for explaining the association of vitamin D, lung development and asthma.73 Previously we investigated the overlap between genes related to vitamin D relative to different DLCGS to infer the role of in utero changes of vitamin D responsive genes in the developing lung and asthma.73 We found the set of vitamin D related genes to be significantly over-represented in DLGCS from independent mouse and human lung development studies, with 38 such genes in both developmental systems. Twelve of 38 (31.6%) were found to be differentially expressed in immortalised B cells derived from 95 subjects with asthma relative to their unaffected siblings.74
Conclusions and next steps
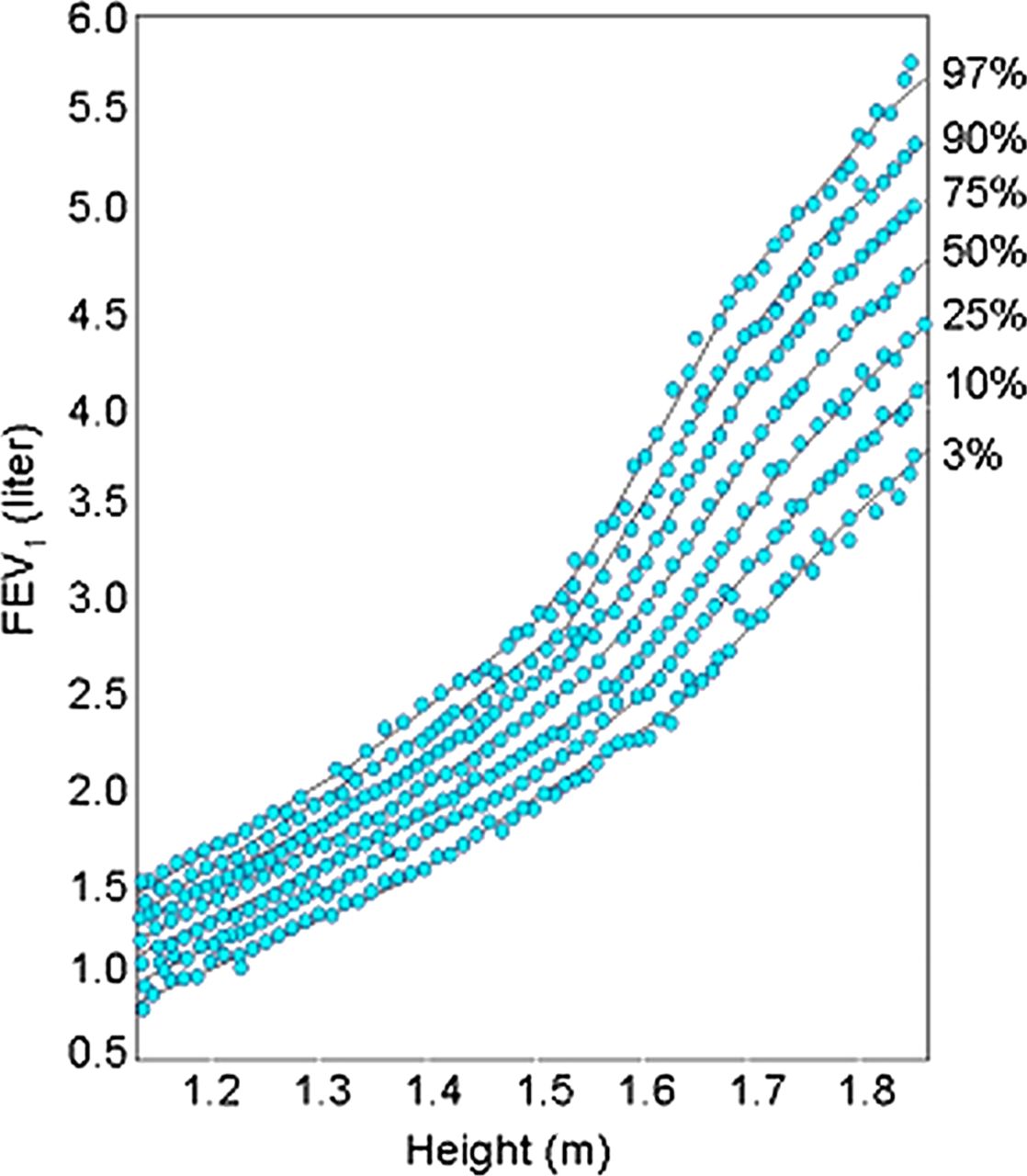
In summary we have shown that the original hypothesis developed by David Barker that fetal development is an important influence on the development of adult disease also extends to childhood asthma. Clearly lung function growth trajectories differ among subjects at 5 years of age, the time in life it can first be measured accurately (figure 4). These differences in developmental trajectories are the result of a complex interaction between environmental factors (IUS, maternal diet, folate, vitamin D and other nutrients, mechanical forces and the large number of genes interacting to build a lung during the developmental process). One of the most important implications of GWAS has been that most of the identified variants associated with common complex diseases are regulatory in function and are active in fetal development, making pleiotropy a critical factor in human complex traits.75 The most important next steps in this research will be to expand our genomic technologies using RNA sequencing on single cell types in development to look at cell–cell interactions, to interrogate a broader range of prenatal human developmental time points, and to develop gene networks to look at the interaction of genes at the different stages of development and how those gene networks are perturbed in disease.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tracking of lung function growth. This figure from the Harvard Six Cities Study77 plots forced expiratory volume in 1 s (FEV1) against height (surrogate for age), with each blue dot representing a repeated measure of FEV1 for a given individual. Lung function tracks along percentile curves over time. Republished with permission from John Wiley & Sons Ltd.
References
Footnotes
-
Contributors SS, KGT, and STW developed the outline for the paper. STW wrote the abstract, Introduction, Conclusions, and next steps. SS wrote Human fetal lung development, Association of lung development genes with asthma and lung function in populations with asthma, IUS in fetal lung development genomics, and Expression quantitative trait loci (eQTL) mapping in development. DC wrote Resources developed to understand the genomic origins of asthma and the Role of folic acid in the development of asthma. ATK wrote The transcriptomic profile of human lung development. LPH wrote the section on vitamin D in lung development.
-
Funding R01 HL097144, R21 HL107927, K08 HL096833, K25 HL091124 and T32 HL007427.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.
-
Data sharing statement This manuscript is subject to the NIH public access policy.
Linked Articles
- Airwaves