Article Text

Abstract
Background There is a strong epidemiological link between smoking and tuberculosis (TB), but the association is confounded by socioeconomic and other factors. A direct relationship between cigarette smoke and poor treatment-related outcomes in patients with TB is therefore questionable. We investigated whether constituents of tobacco smoke impair mycobacterial host immune responses in vitro.
Methodology Preparation of a cigarette smoke extract (CSE) from Marlboro Red cigarettes was standardised and reproducibility verified by mass spectroscopy. Macrophages were derived from peripheral blood monocytes (MDM) and alveolar macrophages from bronchoalveolar lavage fluid from healthy non-smoking volunteers. Mycobacterial uptake (flow cytometric detection of fluorescence using green fluorescent protein-labelled BCG), cytokine responses (ELISA) and mycobacterial containment (colony forming units) was evaluated in both macrophage populations with and without co-culture with CSE, nicotine and a nicotine receptor blocker.
Results Cigarette smoke failed to impair the uptake of mycobacteria by monocyte-derived or alveolar macrophages. CSE (vs no CSE) reduced the mean (SD) BCG-driven macrophage (MDM) interferon γ (IFN-γ), tumour necrosis factor α (TNF-α) and interleukin 10 (IL-10) responses by 56.4 (18.6)%, 67.0 (33.4)% and 77.7 (27.7)%, respectively (p<0.001). Nicotine alone impaired IL-10 and TNF-α production by 48.8 (37)% and 49 (50)%, respectively (p<0.05) through an α-7 nicotine receptor-independent mechanism. In 5-day cultures, CSE impaired mycobacterial (BCG) containment in both monocyte-derived and alveolar macrophages.
Conclusions Cigarette smoke attenuates effector cytokine responses and impairs mycobacterial containment within infected human macrophages derived from the peripheral blood and alveolar compartments, thus supporting the hypothesis that cigarette smoke subverts mycobacteria-related immunity.
- Tuberculosis
- Tobacco and the Lung
- Respiratory Infection
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is the epidemiological association of smoking and heightened risk (approximately double) of tuberculosis (TB) disease and death merely a confounder of socioeconomic or other factors or is it supported by biologically plausible mechanistic data?
What is the bottom line?
-
An animal study has demonstrated higher M tuberculosis colony growth in mice exposed to cigarette smoke but substantial human data are lacking.
Why read on?
-
This study provides novel human data demonstrating several biologically plausible mechanisms by which tobacco smokes subverts human immunity and possibly increases the risk for TB disease.
Introduction
There are an estimated 1.3 billion smokers worldwide.1 Tobacco smoking is the most important preventable cause of death, with an estimated 8% of all adult deaths per year (5 million people) attributable to tobacco smoking. More than 80% of these deaths occur in the developing world.1 One-third of the world’s population is thought to be latently infected with Mycobacterium tuberculosis and an additional 8.8 million new cases are diagnosed with active tuberculosis (TB) each year.2 Several modifiable factors including malnutrition, overcrowding, poverty and HIV co-infection are associated with susceptibility to and spread of active TB.3 In recent years, smoking has been confirmed as another risk factor for TB. Three comprehensive independent systematic reviews and meta-analyses support this association.4–6 Compared with non-smokers, smokers have almost twice the risk of TB infection and of progression from latent to active disease. Smokers are also almost twice as likely to die from active TB.4–6 Based on these data, an estimated 15.8% of TB cases worldwide are probably attributable to tobacco smoking, higher than that attributable to HIV infection (∼11%), alcohol abuse (∼8%) and diabetes (∼7%).7
In the light of these facts, implementation of smoking cessation strategies has been proposed as an important component of TB control programmes in tandem with addressing other modifiable factors such as overcrowding, poverty and alcohol abuse.8 However, in poorly resourced regions, smoking cessation programmes will incur additional cost and place extra demands upon already overburdened clinic staff. It is therefore rightly questioned what priority smoking cessation should receive, particularly because the epidemiological association is weakened by confounding factors such as overcrowding, poverty and alcohol usage, and there is limited experimental data to support the biological plausibility for the association between smoking and TB.9 ,10 In animal models, mice exposed to cigarette smoke for 14 weeks have a significantly higher mycobacterial burden in the lungs and spleen 30 days after challenge with aerosolised M tuberculosis.9 In non-TB models, cigarette smoke has been demonstrated to impair phagocytic function (Staphylococcus, Listeria, Candida) and cytokine responses (lipopolysaccharide, Legionella).11–15 There are no human data on the impact of cigarette smoke extract (CSE) on mycobacterial containment. We hypothesised that constituents of tobacco smoke may attenuate effector cytokine responses and mycobacterial containment in human alveolar and monocyte-derived macrophages (MDM).
Methods
Participants and obtaining macrophages
Healthy HIV-negative non-smoking participants were recruited to provide venous blood samples and/or undergo bronchoscopy and bronchoalveolar lavage. Detailed methods used are provided in the online supplement.
MDM were prepared from peripheral blood mononuclear cells obtained by density sedimentation through Ficoll-Hypaque. The peripheral blood mononuclear cells were seeded at a concentration of 1×106/mL into 24-well plates and allowed to adhere for 6 days. Alveolar macrophages were obtained by bronchoscopy with low-pressure suction using a 300 mL sterile saline lavage. Non-adherent cells were removed at 4 h and appropriate cell concentrations were prepared for each of the experiments performed.
Macrophage infection with BCG
Mycobacterium bovis Bacillus Calmette-Guérin (BCG) expressing green fluorescent protein (BCG-GFP) was used in all infection experiments. MDM were infected with BCG-GFP at a multiplicity of infection of 2:1 and for alveolar macrophages at 2.5:1. MDM were washed with warm phosphate-buffered saline after 18 h to remove non-ingested mycobacteria.
Preparation of CSE and nicotine
A standardised cigarette smoking device was constructed based on the apparatus used in the studies reported by Freed and coworkers (see online supplement). Reproducibility of the extract was assessed using an ABSciex 3200 Qtrap mass spectrometer connected to an Aglient 1200 Series high-performance liquid chromatography. For each experiment, fresh extract was used and added to cultures within 15 min of preparation.
Determination of mycobacterial uptake
Flow cytometric analysis was performed to determine the number of macrophages containing intracellular BCG-GFP. Immediately before acquisition of the cells, 10 μL 7-aminoactinomycin D (7AAD; eBiosciences) was added in order to establish cell viability. Once acquired, the cells were analysed on a FACsCalibur using Cell Quest software.
Cytokine assays
Cytokine concentrations were determined using commercially available ELISA kits and performed according to the manufacturers’ instructions. Triplicates of each experimental condition were prepared and pooled whenever sufficient cells were available. To explore the hypothesis that nicotine can modulate tumour necrosis factor α (TNF-α) production, experiments were conducted using an α7 receptor blocker, α-bungarotoxin (Sigma Aldrich). Macrophages were pre-incubated with α-bungarotoxin-FITC (1.5 μg/mL) for 15 min prior to the addition of nicotine and infection with BCG.
Mycobacterial containment endpoints
To determine the capacity of human macrophages to contain mycobacterial infection, adherent macrophages were infected with BCG and then cultured for 5 days in the presence of 10% CSE. On days 1, 2, 3 and 5, intracellular colony forming units (CFUs) were determined.
Statistical methods
Statistical comparisons were made with the appropriate parametric (t test) and non-parametric tests (Mann–Whitney U test) and, where applicable, paired tests (paired t test or Wilcoxon matched pairs signed rank test). For data involving more than two categories, an analysis of variance (ANOVA) was used (one-way ANOVA or repeated measures ANOVA as appropriate). To correct for multiple comparisons the Tukey test was used. A p value of 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism (V.5.00, GraphPad Software, San Diego, California, USA, http://www.graphpad.com) and OpenEpi (V.2.3.1. http://www.OpenEpi.com, updated 19 September 2010).
Results
CSE and toxicity
The CSE was prepared 26 times over a 9-month experimental period. The mean (SD) concentration of nicotine in the extract was 6.4 (2.6) μg/mL. Exposure of adherent MDM to concentrations of CSE >10% for 24 h showed a dose-dependent reduction in cell viability (see figure E4 in online supplement). Furthermore, at concentrations of ≥20%, spontaneous cell detachment increased (see figure E5 in online supplement). Experiments were conducted using 10% CSE. For uptake and cytokine experiments, CSE was added to the macrophage cultures immediately prior to infection.
Mycobacterial uptake
MDM viability was not affected by BCG infection after 18 h or by co-exposure to either nicotine or cigarette smoke. The mean (SD) percentage viability of uninfected unexposed MDM (determined by 7AAD staining) was 63.3 (5.5)%. Exposure to 10% CSE or 1 μg/mL nicotine did not affect viability (CSE: 67.7 (4.05)%, nicotine: 64.1 (3.8)%; p=0.59). After BCG infection, MDM viability (67.3 (5.8)%) was not significantly different from uninfected macrophage viability (exposed or unexposed) nor from that of infected/CSE-exposed macrophages (68.1 (2.7)%) or infected/nicotine-exposed macrophages (60.9 (9.7)%; p=0.63).
Significant variability in BCG uptake was seen between individuals at 4 h and 18 h. At 4 h BCG uptake was low, but significantly higher in unexposed MDM than in those exposed to 10% CSE (4.5 (2.6)% vs 3.2 (1.6)%; p=0.03). At 18 h, however, no significant difference in uptake was demonstrated between unexposed MDM and those exposed to 10% CSE (24.9 (6.9)% vs 21.3 (6.8)%; p=0.09, figure 1).
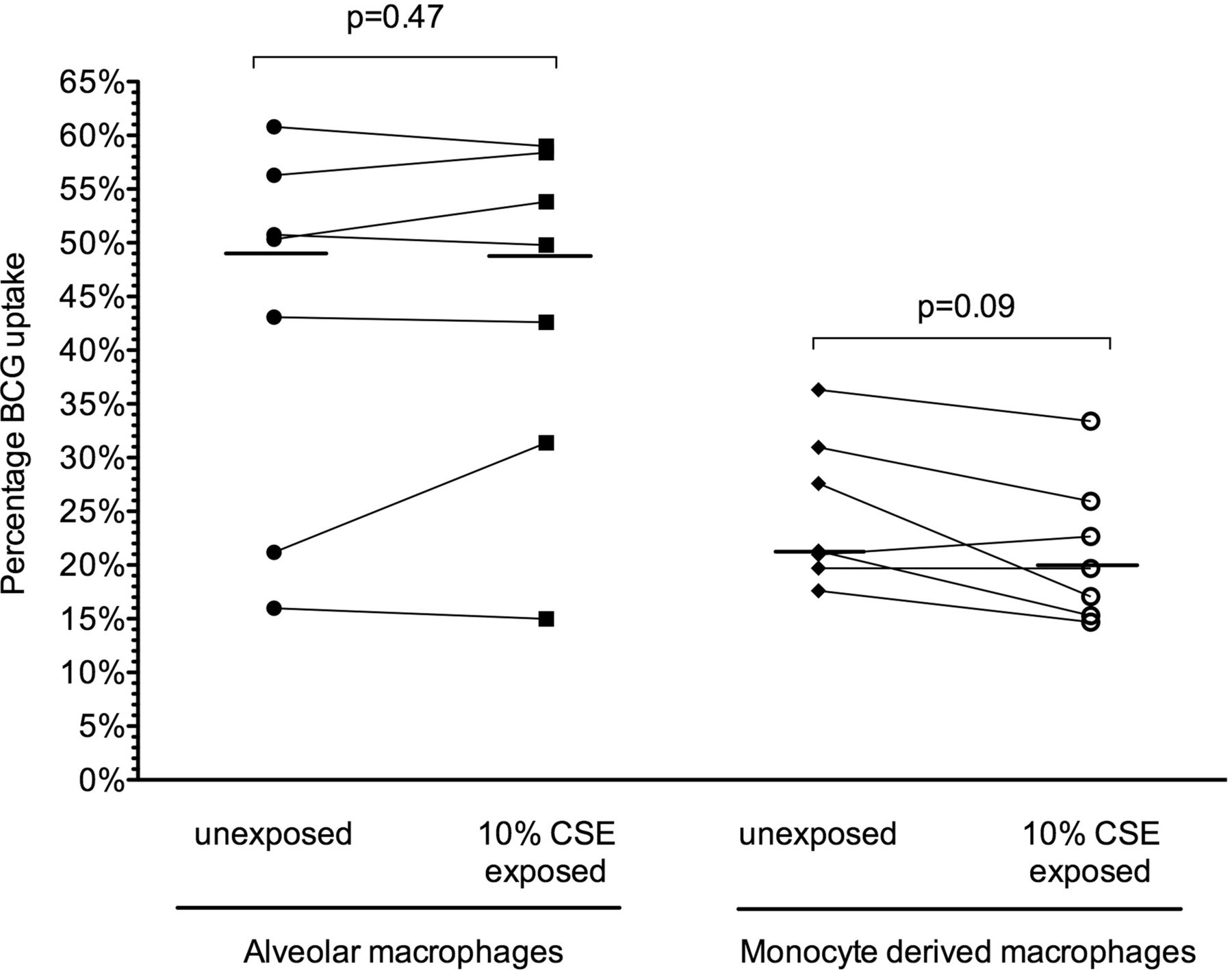
BCG-GFP uptake by alveolar and monocyte-derived macrophages after 18 h. Uptake (percentage of cells positive for GFP by flow cytometry) is depicted for subjects (n=7) following 18 h of infection. Uptake by macrophages with and without exposure to 10% cigarette smoke extract (CSE) from individual donors is depicted by joined lines. The horizontal line represents the median value for all individuals.
Alveolar macrophages exhibited a higher uptake of BCG at 18 h (42.6 (17.4)%), but this was not significantly different from alveolar macrophages exposed to 10% CSE (44.3 (16.1)%) or 1 μg/mL nicotine (35.9 (16.8)%); p=0.28, figure 1). Viability of alveolar macrophages was not affected by infection or co-exposure to 10% CSE or nicotine (see figures E6 and E7 in online supplement).
Cytokine production
IFN-γ production by MDM was measured at 4 h and 18 h after infection. At 4 h the mean (SD) IFN-γ production was minimal (0.05 (0.01) IU/mL) and was not affected by co-exposure to 10% CSE (0.04 (0.18) IU/mL; p=0.43). At 18 h, IFN-γ production by unstimulated MDM as well as those exposed to 10% CSE remained negligible (0.06 (0.03) IU/mL and 0.06 (0.03) IU/mL, respectively; p=1.0). Following BCG infection, a significant increase in mean (SD) IFN-γ was detected (0.28 (0.18) IU/mL). However, macrophages co-exposed to 10% CSE during infection demonstrated significantly less IFN-γ production (0.10 (0.04) IU/mL; p=0.001, figure 2). Production of IFN-γ was confirmed by intracellular staining for IFN-γ and by demonstrating upregulation of IFN-γ mRNA transcription (see figures E8 and E9 and table E2 in online supplement). Due to high basal cytokine production by alveolar macrophages in the 24 h after lavage, cytokine data for alveolar macrophages were not interpretable (data not shown).

Cytokine production at 18 h by monocyte-derived macrophages with and without exposure to cigarette smoke extract (CSE). Each plot represents cytokine production by monocyte-derived macrophages from individual donors. The mean and (SEM) is represented by the horizontal line and error bars. Cells were exposed to BCG for 18 h (BCG-infected); (A) interferon γ (IFN-γ; n=10); (B) tumour necrosis factor α (TNF-α; n=10); (C) interleukin 10 (IL-10; n=8).
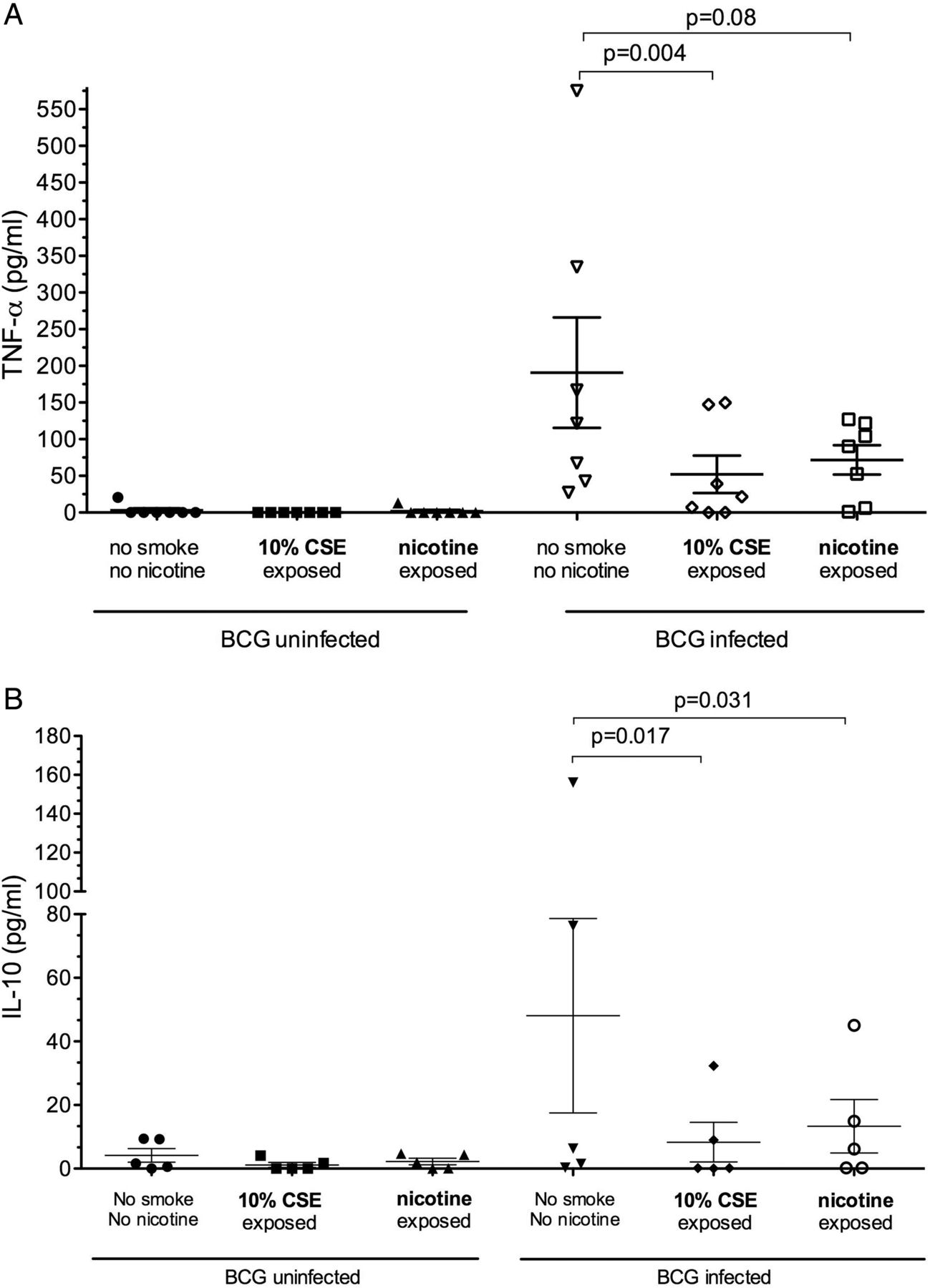
TNF-α production was measured at 18 h after infection with BCG. Similarly to IFN-γ, negligible amounts of TNF-α were secreted by unstimulated and CSE-exposed macrophages (mean (SD) 0.73 (1.8) pg/mL and 1.7 (5.3) pg/mL, respectively; p=0.3). Following infection, TNF-α production increased significantly (137.5 (111.7) pg/mL) but was significantly reduced after co-exposure to 10% CSE (21.63 (45.97) pg/mL; p<0.001 vs control, figure 3).

Effect of nicotine on cytokine production by monocyte-derived macrophages. Each scatter plot represents cytokine production by monocyte-derived macrophages unexposed or exposed to cigarette smoke extract (CSE) or nicotine, and uninfected (left hand panel) or following an 18 h BCG infection (right; BCG-infected). The horizontal bars represent mean (SEM) values. (A) Tumour necrosis factor α (TNF-α) responses (n=7). (B) Interleukin 10 (IL-10) responses (n=5).
Interleukin 10 was measured at 18 h after infection with and without 10% CSE exposure. The results were similar to those for TNF-α and IFN-γ production. Unstimulated and CSE-exposed macrophages produced little or no measurable IL-10 (0.65 (1.65) pg/mL and 0.0 (0.0) pg/mL, respectively). Following infection, IL-10 production was 27.68 (22.5) pg/mL and was significantly reduced by co-exposure to CSE during infection (4.64 (6.75) pg/mL; p<0.001, figure 3).
Compared with the maximal cytokine production of unexposed MDMs, 10% CSE reduced mean (SD) BCG-driven IFN-γ, TNF-α and IL-10 production by 56.4 (18.6)%, 67.0 (33.4)% and 77.7 (27.7)%, respectively. Alveolar macrophage cytokine production and response to CSE or nicotine exposure was not interpretable as basal cytokine production was high when tested 24 h after lavage (data not shown).
Cytokine response to nicotine exposure
Exposure of MDM to nicotine (1 μg mL) alone (as opposed to whole smoke extract) resulted in significantly reduced BCG-driven IL-10 production (40.1 (60.3) pg/mL vs 11.3 (16.4) pg/mL; p=0.03). TNF-α production was similarly reduced but did not reach statistical significance (181.6 (186.1) pg/mL vs 78.7 (52.5) pg/mL; p=0.08, figure 3). The calculated mean (SD) reduction in cytokine production in response to nicotine exposure was 48.8 (37)% for IL-10 and 49 (50)% for TNF-α. Blocking of the nicotine α7 receptor with α-bungarotoxin did not restore TNF-α production by cells exposed to CSE or nicotine (figure 4).
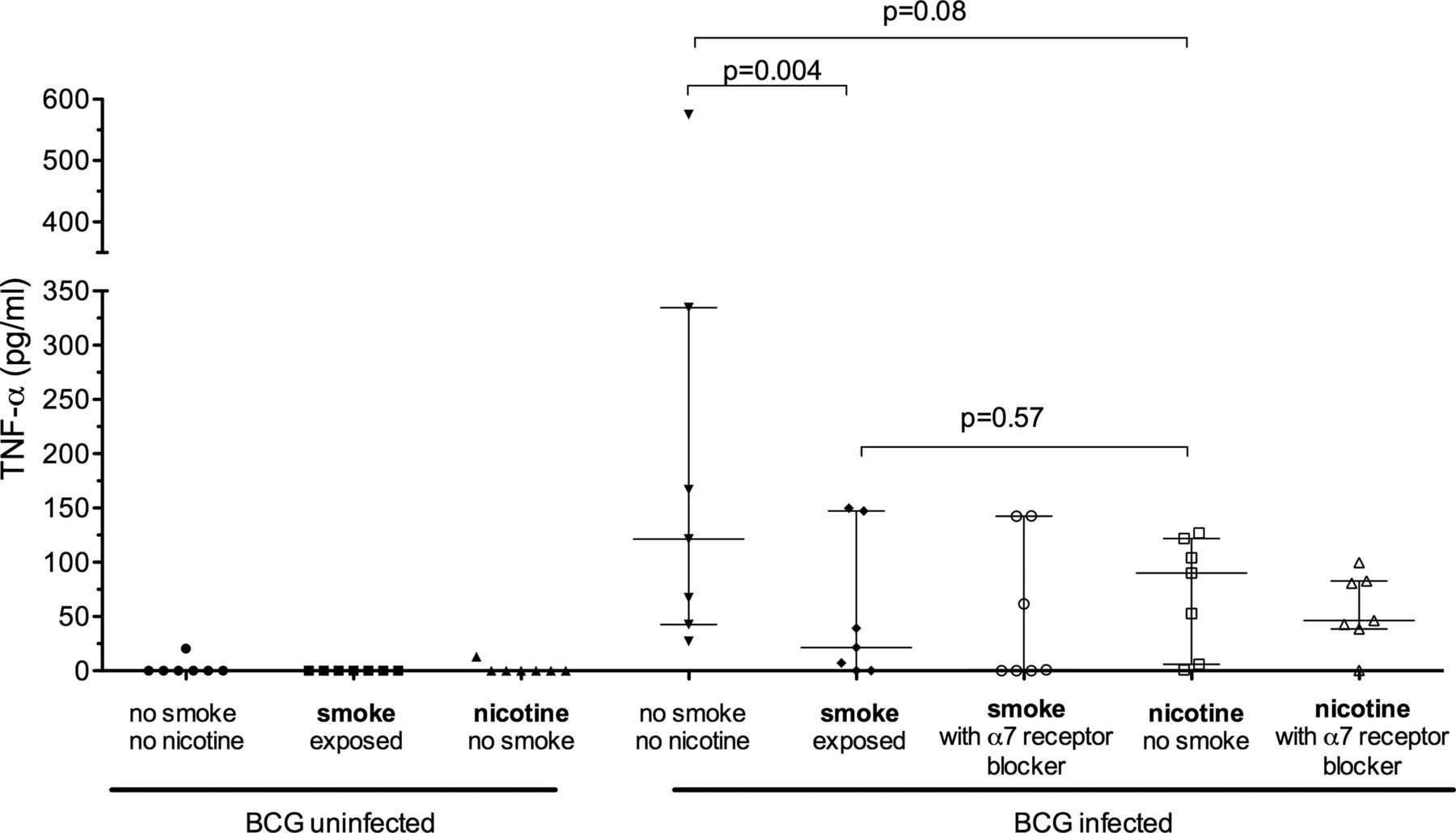
Effect of nicotine on tumour necrosis factor α (TNF-α) production by monocyte-derived macrophages. Each scatter plot represent the production of TNF-α by monocyte-derived macrophages either unexposed or exposed to 10% cigarette smoke extract (CSE) or 1 μg/mL nicotine (n=7). BCG-infected macrophages (18 h) were exposed during infection to CSE/nicotine and a α7 nicotine receptor blocker (α-bungarotoxin). The horizontal and error bars represent median and IQR values.
Mycobacterial containment
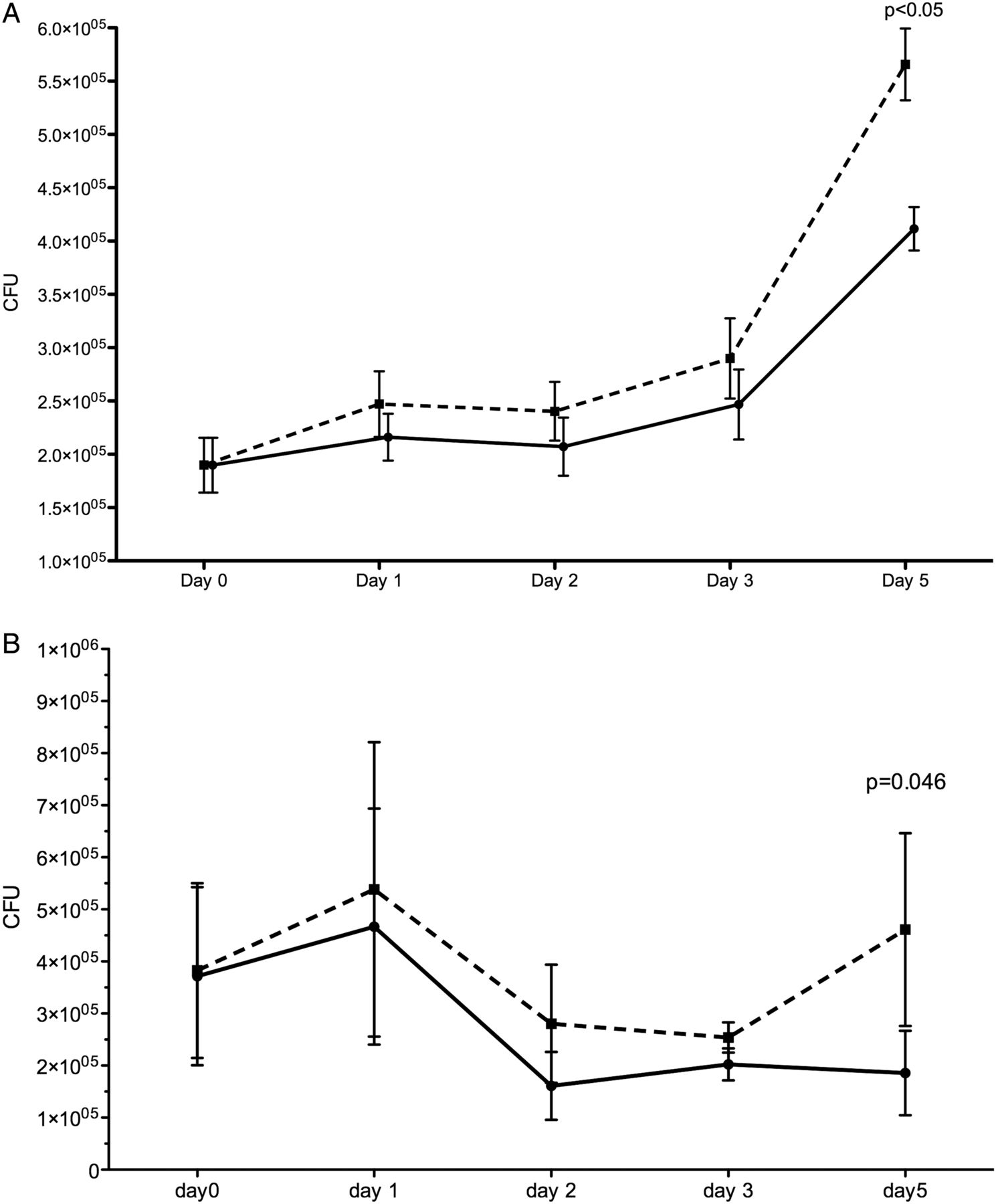
Over a 5-day period, BCG-infected MDM and alveolar macrophages exposed to 10% CSE showed higher CFU counts not accounted for by adherent cell numbers, as the latter did not change (figure 5). At each time point, prior to cell lysis to perform the CFU count, control and CSE-containing wells were inspected under an inverted microscope. No difference in the number and integrity of adherent macrophages could be identified.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serial BCG colony counts over 5 days in (A) monocyte-derived macrophages and (B) alveolar macrophages. Monocyte-derived macrophages and alveolar macrophages infected with BCG were cultured for 5 days after the addition of 10% cigarette smoke extract (CSE) on day 0 (post infection). The solid line represents unexposed macrophages and the dashed line represents CSE-exposed macrophages. Each day represents the time point (post infection) when macrophages were lysed and organism load derived by counting the number of colony forming units (CFU) on solid media. Monocyte-derived macrophages, n=9 (day 1–3) and n=5 (day 5); alveolar macrophages, n=5 (day 1–4) and n=4 (day 5).
Discussion
Our studies of the effect of CSE on the responses of human MDM and alveolar macrophages to mycobacterial infection (BCG) demonstrate that, while the uptake of mycobacteria remained unaffected, the production of key cytokines in the immune response to TB infection (ie, TNF-α, IFN-γ and IL-10) was significantly reduced by exposure to CSE. Furthermore, nicotine alone similarly impaired both IL-10 and TNF-α production, suggesting that it too contributes to this effect. Since the effect of smoke that does not contain nicotine was not examined, it is not clear whether other components of cigarette smoke have a similar effect. We have also demonstrated that both alveolar macrophages and MDM exposed to CSE had significantly higher intracellular bacillary loads after 5 days in culture. The mechanism whereby this occurs and the contribution of other cell types (such as regulatory T cells16) and the observed changes in cellular cytokine release in response to CSE exposure remain unclear and require further study. Collectively, these data provide biological plausibility and support a direct link between smoking and TB, an association which is also confounded by socioeconomic and other factors. To our knowledge, this is the first report of the deleterious impact of CSE on the ability of human macrophages to contain mycobacterial growth. The data strongly support the need for controlled trials about the impact of smoking cessation on TB outcomes.
Intracellular control of mycobacterial replication is a critical step in limiting disease progression.17 Both the virulence of the infecting organisms and the bacterial load affect the ability of macrophages to contain infection.18 ,19 Shang and colleagues demonstrated that smoke-exposed mice had a significantly higher bacterial burden in the lungs and spleen 30 days after infection.9 Additionally, greater numbers of foci of inflammatory cells but reduced influx of CD4 and CD8 effector and memory T cells were present, suggesting impairment in both innate and adaptive responses. However, murine data may not reflect the situation in the human host. To our knowledge, the impact of CSE on the mycobacterial burden in human cells has hitherto not been investigated. In our 5-day human macrophage model, intracellular CFU counts were significantly higher in macrophages exposed to tobacco smoke. We further interrogated several mechanisms that may underpin these observations.
We first investigated the impact of CSE on mycobacterial uptake, about which there are limited data. Using a myelomonocytic cell line (THP-1), Shang and colleagues infected differentiated THP-1 cells with H37Rv during co–exposure to CSE or nicotine. The number of ingested mycobacteria was only measured at 1 h post infection, at which time no difference in uptake was noted.9 Our data, which evaluated several time points, are consistent with these findings and also support the findings of Aldo et al10 where no impairment of BCG-specific phagocytosis by alveolar macrophages was detected. These reports contrast with the effect of tobacco smoke on phagocytosis of organisms other than mycobacteria. Cigarette smoke has been shown to impair macrophage phagocytosis of several organisms including Listeria,11 Haemophilus,20 Staphylococcus,13 Streptococcus21 and Candida.12 The reason for such organism-specific differences remains unclear. Interestingly, Berenson et al,20 in a study using Haemophilus, showed reduced phagocytosis by alveolar macrophages but not MDM from smokers, suggesting functional differences between cells from different compartments. By contrast, we observed no such differences using BCG.
Next we evaluated cytokine responses in human cells after co-exposure to mycobacteria and either tobacco smoke or nicotine. Although there are data for non-mycobacterial stimuli (lipopolysaccharide (LPS), Escherichia coli), there are no data for mycobacterial-specific responses. CSE broadly attenuated cytokine production (IL-10, TNF-α and IFN-γ) from BCG-infected macrophages. This is consistent with the findings of studies in which stimuli other than mycobacteria were used. Ouyang demonstrated reduced TNF-α, IL-β, and IFN-γ in phytohaemagglutinin-stimulated human peripheral blood mononuclear cells following CSE exposure. Similarly, Wewers and Hagiwara, using alveolar lavage cells, showed a reduction in TNF-α (following LPS stimulation) and IFN-γ (following phorbol myristate acetate stimulation), respectively.14 ,15 There are also data about the effect of nicotine on cytokine responses in models other than TB. IL-10 production was impaired in MDM from healthy non-smokers exposed to nicotine patches22 but, in a murine alveolar macrophage cell line infected with Legionella, in contrast to other cytokines, IL-10 was unaffected.23 In our study there was a consistent reduction in IL-10 production but a non-significant reduction in TNF-α production after nicotine exposure. The mechanism underlying these observations remains unclear, but data from non-TB models indicate that smoking may impair signalling through TLR2/424 and several intracellular pathways (NFKB, PI3 K and MAPK).25 ,26 To further interrogate the mechanism underlying our findings, we blocked the α7 receptor. Davies et al,27 based on the work of Wang et al,28 hypothesised that exposure to cigarette smoke may impair TNF-α production through the action of nicotine on the α7 receptor. However, unlike these findings obtained in a mouse model, using an antisense oligonucleotide specific for α7 receptors, we were not able to demonstrate a nicotine α7 receptor-dependent TNF-α response to mycobacterial infection in human cells. Further research is now required to define the mechanisms whereby CSE and nicotine subvert mycobactericidal responses. Such studies should also target whether CSE impacts T-helper cell profiles including regulatory T cells, which may also subvert mycobacterial containment16 and are upregulated in smokers without COPD.29
Our study has several limitations. The model, as a proof of concept, using alveolar and MDMs, BCG-GFP and ex vivo CSE exposure, does not fully represent the lung environment during virulent M tuberculosis infection. BCG-GFP was available at our institution, allowed work to be safely performed outside a BSL3 facility and could be used for all experiments (uptake, cytokine stimulation and containment). It cannot be assumed that the responses to BCG, a non-virulent mycobacterium, that we observed, would be similar for virulent strains of mycobacteria. However, attenuation and subversion of containment of organisms would be expected to be even more pronounced with virulent mycobacteria, and our work provides a theory for how this might occur. Alveolar macrophage cytokine production may have been more informative if assessed after 24 h of culture; however, we chose this point to be consistent across experiments. We did not examine other cytokines such as IL-4, IL-17 that are important in the immune response to TB. We did not, in the first instance, evaluate cells from the blood or lungs of smokers. Smoking alters the white cell count and CD4/CD8 ratios in smokers,30 and the macrophages from smokers’ lungs are structurally different and contain carbon and tar31 resulting in considerable autofluorescence, making the interpretation of flow cytometric results problematic.32 ,33 Given these technical challenges which were borne out by our preliminary experiments, we elected to use cells from non-smokers in the first instance. We did not assess total fluorescence or the uptake of ‘beads’ as a control in our experiments. Other investigators have performed these experiments10; the assessment of phagocytosis by non-receptor mediated uptake was not central to our hypothesis. We did not interrogate specific subfractions of CSE except nicotine and, for reasons of safety, feasibility and complexity, we did not determine whether our findings are applicable to clinical strains of M tuberculosis. Furthermore, whether the concentrations of particulates and soluble constituents of CSE and the duration of exposure in our experiments adequately represent those that occur in the lungs of smokers is difficult to estimate. A variety of exposures have been used in previous studies.13 ,34 ,35 Su and colleagues have proposed that the commonly used CSE of 10% is equivalent to smoking more than one pack of cigarettes per day.35 Nevertheless, the biological significance of the differences in mycobacterial containment seen in our experiments (CSE vs no CSE) remains unclear.
In summary, we have demonstrated that, while not impairing uptake of mycobacteria, exposure to CSE of human macrophages derived both from blood monocytes and the lung attenuates cytokine production and impairs their ability to limit mycobacterial growth. The signalling pathways affected by CSE exposure are yet to be defined, as are the intracellular mechanisms by which mycobacterial growth is facilitated. These data suggest that the suppression of cytokine responses such as IFN-γ and TNF-α in the context of CSE is biologically meaningful and also provides direct evidence for the subversive effect of CSE in mycobacterial infection.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors Conception and design: RNVZ-S, KD, PLS, EDB. Laboratory experiments: RNVZ-S, AB, RM, PLS, AE. Analysis and interpretation: RNVZ-S, PLS, AB, KD, PS, EDB. Drafting the manuscript and important intellectual content: RNVZ-S, PLS, EDB, KD, PS.
-
Funding RNVZ-S was partially supported by a Fogarty International Clinical Research Scholars/Fellows Support Centre NIH grant R24TW007988, South African Thoracic Society and Discovery Foundation Fellowships.
-
Competing interests None.
-
Ethics approval The University of Cape Town Research Ethics Committee granted approval for this study.
-
Patient consent Written informed consent was obtained from all participants prior to enrolment in the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.