Article Text

Abstract
Background Laboratory studies suggest that the clotting cascade is activated in fibrotic lungs. Since humans vary in their tendency to clot due to a variety of inherited or acquired defects, we investigated whether a prothrombotic state increases the chance of developing idiopathic pulmonary fibrosis (IPF) and/or worsens the prognosis of IPF.
Methods We recruited 211 incident cases of IPF and 256 age- and sex-matched general population controls and collected data on medical history, medication, smoking habit, blood samples as well as lung function and high-resolution CT scans done as part of routine clinical care. A prothrombotic state was defined as the presence of at least one inherited or acquired clotting defect or marker of fibrinolytic dysfunction. We used logistic regression to quantify the association between a prothrombotic state and IPF adjusted for age, sex, smoking habit and highly sensitive C reactive protein. Cox regression was used to determine the influence of a prothrombotic state on survival.
Results Cases were more than four times more likely than controls to have a prothrombotic state (OR 4.78, 95% CI 2.93 to 7.80; p<0.0001). Cases with a prothrombotic state were also likely to have more severe disease (forced vital capacity <70% predicted) at presentation (OR 10.79, 95% CI 2.43 to 47.91) and had a threefold increased risk of death (HR 3.26, 95% CI 1.09 to 9.75).
Conclusions People with IPF are more likely to have a prothrombotic state than general population controls and the presence of a prothrombotic state has an adverse impact on survival.
- Idiopathic pulmonary fibrosis
- Interstitial Fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
-
Are people with idiopathic pulmonary fibrosis (IPF) more likely to have a prothrombotic state than general population controls, and does this alter survival among people with IPF?
What is the bottom line?
-
People with IPF were almost five times more likely to have at least one inherited or acquired clotting defect compared with general population controls, and a prothrombotic state in people with IPF was associated with a threefold increased risk of death.
Why read on?
-
This is the largest population-based study to investigate the association between the increased tendency to clot and IPF, which has enabled detailed investigation of the association between individual clotting defects and IPF, disease severity at presentation and survival.
Introduction
Idiopathic pulmonary fibrosis (IPF) is an important public health problem with a disease burden of similar magnitude to ovarian and renal cancers in terms of incidence and survival.1–3 Although there have been recent insights into the pathogenesis of IPF,4 the underlying aetiology of the disease remains poorly understood and there are currently no treatments which improve survival.
Laboratory studies and animal models have suggested that activation of the clotting cascade occurs in fibrotic lungs.5–7 Epidemiological data to support this hypothesis come from a large study using UK primary care data demonstrating that people with IPF are at increased risk of having a venous thromboembolic event (VTE), even before the diagnosis of pulmonary fibrosis was made.8 The association between pulmonary fibrosis and VTE has also been suggested in studies using a large Danish registry9 and American death certificate data.10 A clinical trial in Japan also indicated that anticoagulation in individuals with severe IPF may improve survival.11 A recent placebo-controlled trial of warfarin for IPF was stopped early due to the excessive number of deaths in the warfarin arm that were not due to complications of anticoagulation,12 raising the possibility that manipulation of the clotting cascade using traditional anticoagulation drugs may accelerate rather than treat the disease process.
A significant proportion of the general population are at an increased risk of thrombotic episodes due to a combination of inherited and acquired defects in the clotting cascade.13 ,14 The commonest coagulopathies for people living in the UK are factor V Leiden mutation and elevated factor VIII levels. The aim of our study was to use this natural experiment to investigate the association between clotting dysfunction and IPF and to investigate whether it influenced its natural history. We conducted a case–control study of incident cases of IPF to investigate if people with IPF were more likely to have a prothrombotic state than general population controls, and if this altered subsequent survival.
Methods
Identification and recruitment of cases and controls
We identified potential incident cases of IPF seen at five teaching hospitals and eight district general hospitals in the Greater Trent region and Wales between January 2010 and February 2012 (see figure 1 and online supplementary appendix 1).
Controls were frequency matched on age and sex, and consisted of people in the general population who did not have a diagnosis of IPF, and were identified from 10 primary care centres involved in the UK Primary Care Research Network in the Greater Trent Region.
Data collection
Cases and controls were invited to participate in the study by their respiratory physician or general practitioner. All participants were sent a questionnaire asking for details of medical history, medication and smoking habit and had a venous blood sample taken. We also collected results of pulmonary function tests conducted according to the American Thoracic Society/European Respiratory Society (ATS/ERS) task force guidelines15–17 and high-resolution CT (HRCT) scans done as part of routine care for the cases. All participants were tagged with the NHS Information Centre to enable us to collect data on subsequent mortality and cause of death.
Laboratory analysis
Laboratory testing was conducted for inherited clotting defects including factor V Leiden mutation, prothrombin G20210A mutation, methylenetetrahydrofolate reductase deficiency, antithrombin III deficiency, elevated factor VIII levels, protein C and free protein S deficiency, acquired clotting defects including lupus anticoagulant and anticardiolipin antibodies and markers of fibrinolysis including D-dimer levels and clot lysis times. All laboratory analyses were performed in the laboratories at Nottingham University Hospitals NHS Trust in accordance with standard clinical laboratory practice.
Statistical analysis
We only included cases with diagnoses of definite or probable usual interstitial pneumonia (UIP) in the main analyses. Our main exposure was a prothrombotic state, which was a composite binary variable defined as having at least one of the measured inherited or acquired clotting abnormalities.
Logistic regression was used to generate ORs for the association between a prothrombotic state and IPF adjusting for age category and sex, which was our baseline statistical model. We investigated the association of the individual clotting defects stated above and IPF by replacing our composite prothrombotic state variable with the individual clotting defect in turn. We repeated the analyses, also adjusting for smoking habit and highly sensitive C reactive protein (hsCRP) as potential confounding variables. Using the same baseline and multivariate models, we looked for an exposure-response signal in terms of the absolute number of clotting defects present and IPF, and a quantitative analysis on the most common clotting defects and markers of clotting dysfunction and IPF after excluding individuals currently on warfarin. We conducted a separate analysis among the cases to determine if a prothrombotic state was associated with pulmonary function indices of disease severity. The main analyses were repeated stratifying cases by radiological diagnoses of definite or probable UIP. Cases with fibrotic non-specific interstitial pneumonitis (NSIP) and unclassified fibrotic lung disease were analysed separately. Cox regression modelling was used to investigate if a prothrombotic state was associated with increased mortality among people with IPF using the same statistical models. We assigned the date participants gave consent to enter the study as the start date, and the date of death or last data collection as the stop date. We assessed the impact of the same coagulation defects and the association between the absolute number of clotting defects present on survival. The analyses were repeated adjusting for baseline forced vital capacity (FVC) and carbon monoxide transfer factor (Tlco) percentage predicted. Proportional hazard assumptions were checked using log–log survival plots and scaled Schoenfeld residuals.
The study was designed to include 244 cases of IPF and 244 controls frequency matched to age and sex to provide more than 90% power to detect an OR of ≥2 on the assumption that the prevalence of thrombophilia in the control group was 20%. STATA V.11.0 was used for all statistical analyses and hypothesis testing.
Results
Case–control analysis
We recruited 306 incident cases of physician-diagnosed IPF and 256 controls during the study period. Following central review of HRCT scans by two thoracic radiologists (KP and MK), 110 cases of definite UIP and 101 cases with probable UIP remained (table 1). Only these 211 cases of definite or probable UIP were included in subsequent analyses. The mean (SD) age of cases with definite or probable UIP was 73.7 (8.5) years and the cases were predominantly male. Cases were more likely than controls to have a previous thromboembolic event, history of ischaemic heart disease, to be currently or previously on aspirin and/or warfarin and to have a hsCRP level of >3 mg/mL (table 2).
Radiological diagnoses within the cohort of patients with physician-diagnosed IPF
Demographics of incident cases of idiopathic pulmonary fibrosis (IPF) and general population controls
A prothrombotic state was more than four times more common in cases than in controls in both our baseline model (OR 4.38, 95% CI 2.85 to 6.74; p<0.0001) and our multivariate model (OR 4.78, 95% CI 2.93 to 7.80; p<0.0001) (table 3). After excluding individuals currently on warfarin, cases (n=194) were more than seven times more likely than controls (n=250) to have factor VIII levels above 165 IU/dL in both the baseline model (OR 7.20, 95% CI 4.65 to 11.18; p<0.0001) and the multivariate model (OR 7.02, 95% CI 4.27 to 11.51; p<0.0001). There was no effect modification by age or sex on any of our findings.
Association between individual clotting defects and IPF
Exposure-response analyses, association with disease severity and radiological diagnosis
Cases were more than five times more likely to have two or more clotting defects than controls in both the baseline model (OR 7.66, 95% CI 2.81 to 20.88) and the multivariate model (OR 5.85, 95% CI 2.05 to 16.81). We found evidence of an exposure-response signal with increasing levels of factor VIII, D-dimer and clot lysis times (table 4).
Number of clotting defects and exposure-response relationship of common clotting abnormalities*
Pulmonary function test results at time of diagnosis were available for 200 of the 211 subjects (95%) in our IPF cohort. The mean (SD) FVC percentage predicted was 84.4 (19.6) and mean (SD) Tlco percentage predicted was 45.4 (16.4). We found an association between an increased tendency to clot and severity of IPF at presentation (table 5).
Risk of a prothrombotic state* in people with IPF stratified by pulmonary function indices
The increased tendency to clot was marginally higher among cases with definite UIP (n=110) in the baseline model (OR 5.77, 95% CI 3.21 to 10.34; p<0.0001) and in the multivariate model (OR 6.31, 95% CI 3.32 to 11.97; p<0.0001). Cases with probable UIP (n=101) had a threefold increase in tendency to clot in both the baseline model (OR 3.37, 95% CI 1.98 to 5.74; p<0.0001) and the multivariate model (OR 3.49, 95% CI 1.90 to 6.40; p<0.0001). We also found that the increased tendency to clot was present in the subset of cases with fibrotic NSIP (n=28) in the baseline model (OR 4.74, 95% CI 1.69 to 13.33; p=0.001) and the multivariate model (OR 5.71, 95% CI 1.82 to 17.97; p=0.001) or unclassifiable fibrotic lung disease (n=67) in the baseline (OR 4.29, 95% CI 2.22 to 8.27; p<0.0001) and multivariable models (OR 4.05, 95% CI 1.97 to 8.32; p=0.0001).
Survival analysis
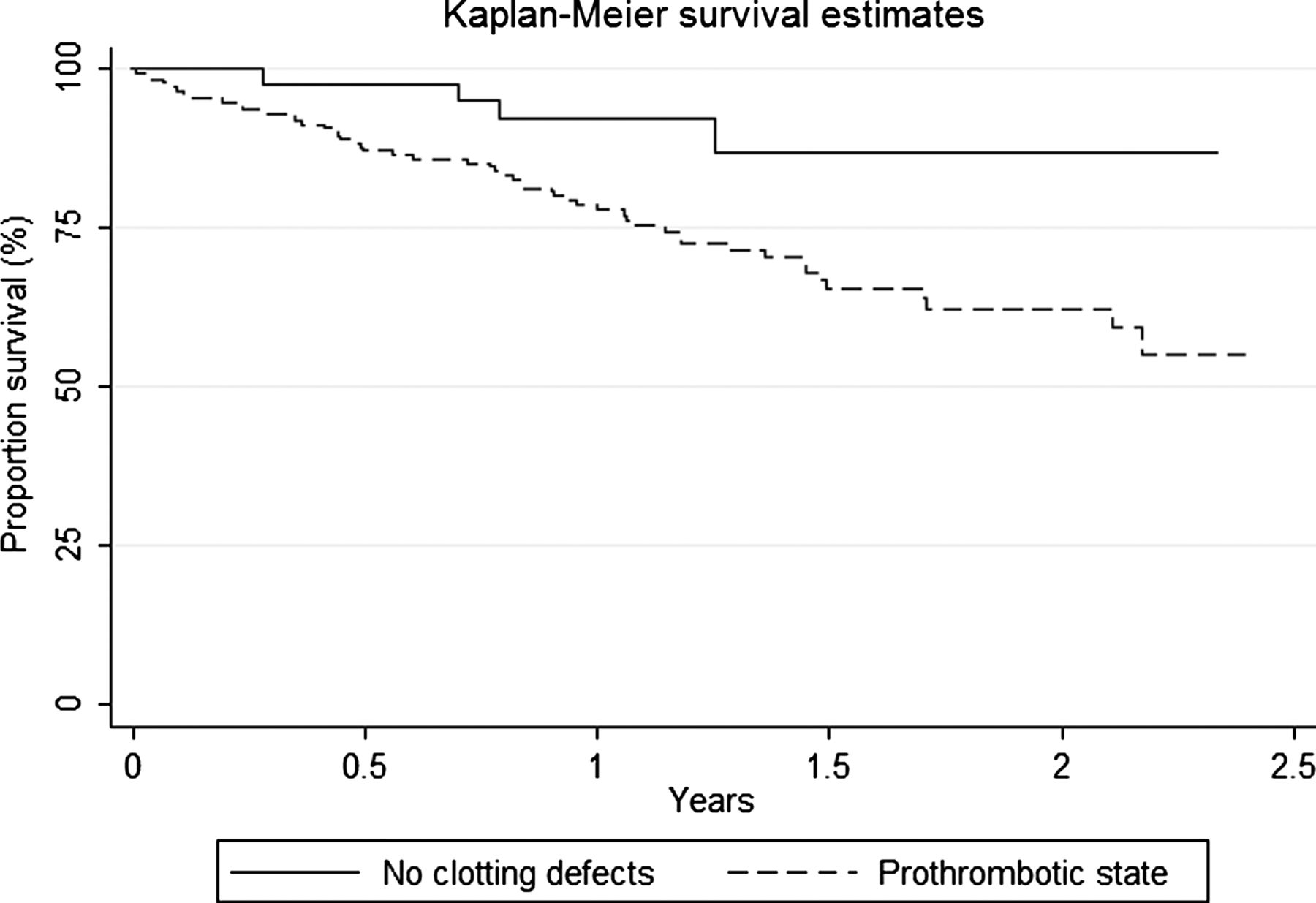
The median follow-up for people with definite or probable UIP after the start date was 1.14 years. During this period, 56 of the 211 (26.5%) individuals with IPF died. The overall mortality rate of the cohort was 215.0 (95% CI 165.5 to 279.4) per 1000 person-years. A prothrombotic state increased mortality by three times among people with IPF after adjusting for age and sex (HR 3.38, 95% CI 1.22 to 9.21; p=0.006) and after adjusting for age, sex, smoking habit and hsCRP (HR 3.45, 95% CI 1.15 to 10.65; p=0.007), with a fourfold increased risk of death among cases with two or more clotting defects (table 6, figure 2). The increased mortality was unchanged after adjusting for baseline FVC and Tlco (HR 3.50, 95% CI 1.09 to 9.75; p=0.015). Current anticoagulation with warfarin or hsCRP level >3 mg/mL had no association with survival (table 7). There was no evidence of effect modification by age or sex, or that the proportional hazards assumptions for our final models were not met (p=0.425; scaled Schoenfeld global test).⇓
Cox regression modelling of individual clotting defects and survival in the IPF cohort
Association between demographic characteristics and survival in the IPF cohort

Flow chart depicting recruitment of incident cases of IPF and general population controls.
{kind=link}
{kind=link}
Kaplan-Meier plot of survival amongst individuals with IPF with and without a prothrombotic state.
Discussion
In this large population-based study we found that a prothrombotic state was almost five times more common in people with IPF than in general population controls. People with IPF were also more likely to have multiple clotting defects. We also found that a prothrombotic state is associated with disease severity at diagnosis measured by pulmonary function indices, and carries a threefold increase in mortality.
The main strength of our study is its large size of well-phenotyped individuals with IPF and the ability to study the association with individual clotting defects. Cases were recruited from specialist centres and district general hospitals, which enabled us to capture the full spectrum of patients with IPF and avoid sampling bias that occurs with studies conducted only at tertiary referral centres. We are confident of the diagnosis of IPF within our cohort as all HRCT scans were reviewed by two thoracic radiologists who are part of the regional interstitial lung disease multidisciplinary team, and cases were stratified into definite or probable UIP based on the ATS/ERS diagnostic criteria for diagnosis of IPF.18 Furthermore, we have previously shown good interobserver agreement (κ=0.67) between these two thoracic radiologists.19 The age distribution and survival of individuals with IPF in our study is similar to other population-based cohorts of IPF,20–24 reassuring us of the robust diagnosis of IPF and making our findings generalisable. Our main exposure was a prothrombotic state confirmed on laboratory testing, ensuring that there was no recall bias. Furthermore, using the higher baseline value of levels >165 IU/dL as our definition of elevated factor VIII reduced the possibility of misclassification of a procoagulant state in this older population. As we only included incident cases of IPF, the findings from our cohort analysis are unlikely to be due to survival bias. In addition to this, the detail of information collected enabled us to explore and adjust for various confounding factors including systemic inflammation and to investigate the possibility of effect modification.
Although our data are robust, this study did have a number of factors that need consideration. Only five out of 306 (1.6%) in our cohort of people with physician-diagnosed IPF underwent a lung biopsy, which is in keeping with current clinical practice in the UK, making our data generalisable to this population but less so to other studies that used a histological diagnosis of UIP. However, current evidence suggests a high specificity for the diagnosis of IPF made on HRCT scanning in comparison with surgical lung biopsy,24–27 making it likely that any diagnostic imprecision, if present, is minimal. In the subgroup of 110 people with definite UIP on HRCT, the risk of having a prothrombotic state compared with general population controls was markedly increased. Furthermore, a study by Fell and colleagues has suggested that age is a strong predictive factor of IPF in people with an atypical UIP radiological appearance, whereby individuals over the age of 65 years had 89% specificity for confirming IPF by surgical lung biopsy.23 For the subgroup of cases in our study with probable UIP (n=101), approximately 85% were over the age of 65, reassuring us that the majority of these cases have a high certainty of having UIP. The boundaries between the disease entities such as UIP and NSIP within lung fibrosis still remain unclear,23 and this is highlighted by studies showing that familial IPF has a wide clinical phenotype28 and that people may have different histological findings in different regions of their lungs.24 This may explain why our findings indicate that people with fibrotic NSIP and unclassifiable lung disease also have an increased tendency to clot.
Another factor for consideration is that some of the coagulation proteases and markers of clotting dysfunction also act as acute phase response proteins, raising the possibility that some of our findings could be attributed to inflammation or intercurrent infection. However, after adjustment with hsCRP, there was still a strong association that a prothrombotic state was more common in people with IPF compared with controls and is unlikely to be due to residual confounding. Our main exposure variable is a prothrombotic state, as defined by the British Committee for Standards in Haematology.13 This was a composite binary variable defined as having at least one inherited or acquired clotting abnormality, assuming each coagulopathy to have an equal effect on the prothrombotic state. However, although this approach is useful from a statistical perspective, it needs to be considered that different coagulopathies may vary in magnitudes of clotting tendency.
The number of deaths in our IPF cohort is high given the short follow-up period, but our cumulative survival estimates are similar to other observational IPF studies20–22 ,24 and show a significant difference in survival in people with IPF with and without a prothrombotic state. Our study found no association between survival in people with IPF and being currently on warfarin. However, given the small number of deaths in the cohort and lack of statistical power to address this question, no firm conclusions can be drawn.
We used people from the general population without a diagnosis of IPF as our control group as this is a representative sample of individuals without IPF taken from the population from which the cases arose. Because all people with IPF are diagnosed in hospital, the risk of selection bias by recruiting controls from primary care centres is minimal. Although the response rate from our controls was substantially lower than the cases (28% vs 86%), we believe that our findings are representative of the general population, as potential controls approached were not aware of the hypothesis tested or of their individual thrombotic state.
The final number of cases with definite or probable UIP (n=211) included in the final analyses falls slightly short of the sample size estimated by the power calculation (n=244). However, this was on the assumption that a prothrombotic state would be present in 20% of the controls. We found that the prevalence of a prothrombotic state was significantly higher in our controls (46.5%), therefore providing adequate power with a reduced sample size.
To our knowledge, this is the first population-based study to investigate the association between a prothrombotic state and IPF. While the findings of our study confirm this association, it does not establish causality. Possible explanations for our findings include a prothrombotic state being a risk factor for developing IPF, or the possibility that IPF leads to the development of a prothrombotic state, or that they share risk factors. The findings of this study are consistent with epidemiological and laboratory studies suggesting that activation of the coagulation cascade within the lung may be involved in the pathogenesis of IPF. Using a large UK primary care database, we previously demonstrated that people with IPF were more likely than controls to have a VTE, even in the time period before the diagnosis of IPF was made,8 and this observation is supported by our data from the current study where current anticoagulant therapy and coronary heart disease were associated with IPF. More recently, a study using registered death certificates in the USA showed that thromboembolic events were higher in people with pulmonary fibrosis than in those with chronic obstructive pulmonary disease or lung cancer and the general population.10 Furthermore, this study also suggested that individuals with pulmonary fibrosis and a VTE died at a younger age than those with pulmonary fibrosis alone.10 Using a large registry of Danish patients, Sode and colleagues found that the risk of developing idiopathic interstitial pneumonia (IIP) was higher in people who had previously had a VTE, and this risk was highest among the subset of people who had not received anticoagulant treatment.9
Further evidence that the clotting cascade is involved in the aetiology of IPF comes from laboratory studies and animal models. In vivo studies have shown that fibrinolytic activity is suppressed within lungs of people with IPF, resulting in fibrin deposition in the interstitial and alveolar spaces,29 and there is evidence of high levels of tissue factor in bronchoalveolar lavage fluid of people with IPF30 as well as overexpression of plasminogen activator inhibitor-1 (PAI-1) in type 2 pneumocytes within honeycomb lesions,31 all of which may increase local fibrin deposition. Patient studies and animal models of fibrotic lung disease also support the role of thrombin, which plays a central role in the final common pathway of the clotting cascade in activating protease-activated receptor type 1 (PAR-1) expressed in lung tissue.32–34 Studies have demonstrated that PAR-1 is highly expressed on cells implicated in fibrosis in the lungs of people with IPF,32 suggesting the receptor plays an important role in the pathogenic process. Activation of PAR-1 receptors leads to the induction and release of potent profibrotic35 and proinflammatory mediators,5 induces differentiation of fibroblasts to myofibroblasts36 and promotes extracellular matrix protein production.37 Furthermore, active inhibition of the clotting cascade using nebulised heparin, urokinase or activated protein C reduces collagen accumulation and the development of fibrotic lesions in experimental models of lung fibrosis.38 ,39
There have been mixed results of clinical trials using anticoagulants as potential treatments for IPF. A clinical trial in Japan using warfarin and corticosteroids or corticosteroids alone in individuals with progressive IPF indicated that anticoagulation may prolong survival.11 This is different from the findings of the anticoagulant effectiveness in IPF (ACE-IPF) trial which was stopped due to an excessive number of deaths in the warfarin arm (14 warfarin vs 3 placebo; p=0.005). The majority of deaths in the warfarin arm were due to respiratory failure,12 suggesting that manipulation of the clotting cascade with warfarin may lead to acceleration of lung fibrosis. While disappointing, this provides further evidence that the clotting cascade may be central to the disease process in IPF and that the coagulation cascade remains a valid therapeutic target.
In summary, our findings suggest that people with IPF are more than four times more likely to have a prothrombotic state and almost six times more likely to have two or more clotting defects compared with general population controls. The increased tendency to clot is also associated with disease severity at presentation and increases mortality by threefold among people with IPF. These findings demonstrate strong differences in clotting dysfunction and firmly establish the clotting cascade at the centre of the aetiology and prognosis in IPF. Studies from animal models have suggested that manipulation of the clotting cascade can alter the course of fibrosis, raising the possibility that manipulation of the clotting cascade could be an important treatment avenue for people with IPF. However, the recent placebo-controlled trial of warfarin in IPF has demonstrated that broad-spectrum anticoagulation may not be the answer, and further mechanistic work into how best to manipulate the clotting cascade to improve the outlook for people with IPF is warranted.
Acknowledgments
The authors would like to thank the following people for their help: Dr K Amsha (Kingsmill Hospital), Dr R Berg (Royal Derby Hospitals), Professor MK Whyte (Sheffield Teaching Hospitals), Dr S Agrawal (Glenfield Hospital), Dr T Rogers (Doncaster Royal Infirmary), Dr U Nanda (Queen's Hospital), Dr J Hadfield (Chesterfield Royal Hospital), Dr J Mann (Royal Wolverhampton Hospitals), Dr I Le Jeune (Queen's Medical Centre), Dr G Saini and Mrs J Morgan (Nottingham City Hospital), Dr D Boldy (Pilgrim Hospital), Dr S Matusiewicz (Lincoln County Hospital), Dr P Floodpage (Royal Gwent Hospital), Dr B Hope-Gill (University Hospital Llandough), Dr K Harrison (Morriston Hospital), Dr Fraser (East Leake Medical Group), Dr Loveland (Wheatbridge Health Centre), Dr Saunders (Dronfield Medical Centre), Dr Grant (Darley Dale Medical Centre), Dr Perkins (Perkins, Bawtry and Blyth Medical Centre), Dr Paak (Lindum Medical Practice), Dr Quereshi (The Heath Surgery), Dr Latham (Birchwood Medical Practice), Dr Effingham (Bridgegate Surgery), Dr Meer (Tall Tree Surgery), Mrs S O'Malley (Trent Comprehensive Local Research Network), Professor T Avery (University of Nottingham), Miss N Bailey-Flitter and Mr C Beecroft (Trent Primary Care Research Network), Mr K Hines and Mr P Thorpe (Department of Radiology, Nottingham University Hospitals NHS Trust), Mr R Kirby and Mr B Gordon (Department of Haematology, Nottingham University Hospitals NHS Trust), Mr S Martin (Department of Immunology, Nottingham University Hospitals NHS Trust), Mr N Grieg (Department of Clinical Chemistry, Nottingham University Hospitals NHS Trust), Lung Function and Sleep Services Units of all the participating hospitals, Mr G Meakin, Miss H Bailey, Miss R Simms, Miss F Fordham, Mrs S Churchill, Mrs D Barber, Mr G Hearson and Mrs S Ledger (Nottingham Respiratory Research Unit), Dr Ian Johnston and the members of the Trent Lung Fibrosis Steering Group, the Medical Research Council and all the participants of the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors AWF, TM and RBH conceived and designed the study. VN, NT, KP and MK were involved in the acquisition of the data. VN, AWF, TM and RBH were involved in the analyses of the data. VN, AWF, TM, NT, GJ, SRJ, GD, KP, MK and RBH were involved in the interpretation of the data and in writing or revising the paper before submission. VN takes responsibility for the integrity of the work in this paper and is the guarantor of the paper.
-
Funding This study was funded by the Medical Research Council (G0802065/1). RH is funded by the GSK/BLF chair of Epidemiological Respiratory Research.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethical approval for the study was granted by the Nottingham Research Ethics Committee (REC reference 09/H0403/59) and research and development approval was obtained for each participating centre.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Airwaves