Article Text

Abstract
Introduction The burden of chronic heart failure (HF) is rising owing to an increased survivorship after myocardial infarction (MI). Pulmonary structural remodelling in patients with HF may protect against oedema while causing dyspnoea, the predominant symptom associated with HF. The cellular and molecular mechanisms underlying these processes in HF are poorly understood. We hypothesised that pulmonary venous hypertension (PVH) following MI provides a mechanical stimulus for structural remodelling of the lung via monocyte chemoattractant protein-1 (MCP-1).
Methods Human lung microvascular endothelial cells (HLMVEC) and Ea.Hy 926 cells exposed to cyclic mechanical strain (CMS) in vitro were analysed for MCP-1 expression and activation of signalling intermediates. HF was induced in Sprague–Dawley rats 16 weeks after MI; a cohort was rescued with AAV9.SERCA2a gene therapy to reduce PVH.
Results HLMVEC and Ea.Hy 926 cells exposed to CMS upregulated MCP-1 gene expression and protein release in an extracellular-signal-regulated kinase (ERK) 1/2 dependent manner. Supernatants from these experiments stimulated fibroblast (human fetal lung fibroblast -1) and pulmonary artery smooth muscle cell proliferation and differentiation. Total lung collagen, a marker of structural remodelling, and MCP-1 gene expression were increased in the lungs of rats with post-MI HF. SERCA2a gene therapy that attenuated PVH after MI was associated with lower levels of lung collagen and MCP-1 gene expression in the lung.
Conclusions Mechanical strain associated with PVH may stimulate pulmonary structural remodelling through ERK 1/2 dependent induction of MCP-1. These findings provide insights into the pathophysiology of lung remodelling in HF and highlight novel, potential therapeutic targets.
- Systemic disease and lungs
- Pulmonary oedema
Statistics from Altmetric.com
Key messages
What is the key question?
Chronic heart failure (HF) causes vascular and structural remodelling in the lung, which in turn contributes to dyspnoea: the mechanisms underlying this pulmonary remodelling are unknown.
What is the bottom line?
In a rodent model of HF, the lung produced the chemokine monocyte chemoattractant protein-1 (MCP-1) when subjected to mechanical strain from pulmonary venous hypertension and haemodynamic improvement after SERCA2a gene was associated with decreased pulmonary MCP-1 and collagen content.
Why read on?
This is the first study to show that mechanical strain applied to lung microvascular endothelial cells can stimulate MCP-1 release and that gene therapy in HF rodents can reduce lung-derived MCP-1 and potentially reverse structural remodelling.
Introduction
The burden of chronic heart failure (HF) is rising owing to an increased incidence of, and improved survival after, myocardial infarction (MI).1 ,2 Dyspnoea is a common and debilitating symptom; 37% of patients with HF had shortness of breath on moderate exertion or at rest.3 Pulmonary microvascular permeability and gas transfer was reduced in a cohort of patients with HF even after effective treatment of pulmonary oedema,4 ,5 implicating a structural abnormality of the alveolar-capillary membrane.6 Fibrotic changes in the pulmonary circulation (vascular remodelling) and alveolar walls (structural remodelling) both contribute to functional limitation by reducing diffusing capacity and lung compliance.7 ,8 Structural remodelling of the lungs in HF is characterised by an increased number of myofibroblasts with excess collagen and reticulin deposition in alveolar septae;9 ,10 however, the mechanisms underlying these processes in HF are poorly understood.
Proinflammatory cytokines (eg, interleukin (IL)-6 and tumour necrosis factor α (TNF-α)) contribute to cardiac dysfunction and progression of HF, but chemokines (IL-8 and monocyte chemoattractant protein (MCP)-1), adhesion molecules, nitric oxide and endothelin may also play a significant role.11–13 Elevated circulating levels of both cytokines (IL-6, TNF-α) and chemokines (IL-8, MCP-1) correlate with disease progression14 ,15 regardless of the aetiology of HF, suggesting a potential, common pathogenic pathway initiated by HF itself rather than its causes.16
The source of proinflammatory and profibrotic mediators in HF is unknown. Proposed mechanisms include direct release from damaged myocardium and bacterial endotoxin translocation across an oedematous gut wall.17 The lung is an alternative source: IL-6 and MCP-1 (chemokine (C-C motif) ligand (ccl)-2) gene expression are increased in the lungs of animals with experimentally induced HF, IL-6 having been localised to the endothelium.18 The lungs are immediately upstream of the failing left ventricle, which causes pulmonary venous hypertension (PVH) and hence mechanical strain on the affected vessels. Endothelial cells (EC) are known to be ‘mechanosensitive’, for example, human lung microvascular endothelial cells (HLMVEC) expressed metalloproteinase-2 in response to mechanical strain.19 Other EC released MCP-1, intercellular adhesion molecule-1 and IL-8 in response to strain.20 ,21 We, therefore, hypothesised that raised left atrial pressure (LAP) secondary to HF post-MI imparts a mechanical strain at the pulmonary microvasculature, leading to the release of mediators, including MCP-1, that contribute to alveolar capillary membrane remodelling.
Sarco(endo)plasmic reticulum calcium ATPase 2a (SERCA2a) is an enzyme that transfers calcium to the sarcoplasmic reticulum (SR), underpinning decay of the calcium transient and cardiomyocyte relaxation during diastole. SERCA2a expression and activity is reduced in the failing myocardium with decreased SR calcium uptake.22 ,23 SERCA2a gene transfer increased survival and attenuated changes in left ventricular end diastolic pressure (LVEDP) in rat models of HF,24–26 thereby providing an opportunity to study the effects of PVH in relative isolation.
In this study, we investigated the role of mechanical strain in the production of MCP-1 by EC and the effect of this on pulmonary remodelling. Furthermore, we determined the effects of SERCA2a gene therapy on pulmonary expression of selected mediators and pulmonary remodelling in an ischaemic model of HF.
Methods
Cell culture
HLMVEC (Lonza, Wokingham, UK), maintained in complete EC growth medium microvascular 2, were used between passages 3 and 8. The human EC line (Ea.Hy 926) and human fibroblast cell line (human fetal lung fibroblast (HFL)-1) were purchased from the Centre for Disease Control and Prevention (CDC, Atlanta, USA). Ea.HY 926 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine, 10% fetal calf serum (FCS, heat inactivated), 100 U/mL penicillin, 100 μg/mL streptomycin, 100 μM hypoxanthine, 0.4 μM aminopterin and 16 μM thymidine (Sigma-Aldrich, Poole, UK). Pulmonary artery smooth muscle cells (PASMC) (Lonza, Wokingham, UK) were maintained in DMEM supplemented with 15% FCS, 2 mM glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin.
Mechanical strain
HLMVEC and Ea.Hy 926 cells were seeded onto collagen-coated BioFlex 6 well plates (DunnLab, Asbach. Germany), grown to confluence and serum restricted for 16 h (0.1% FCS). The medium was refreshed immediately prior to application of the cyclic mechanical strain protocol (CMS: 30% elongation, at 0.3 Hz, for 4 h) driven by the Flexercell FX 4000 strain unit (DunnLab, Asbach, Germany). To determine the roles of p42/44 mitogen-activated protein kinase (MAPK) in strain-induced MCP-1 expression, cells were pretreated with MEK 1/2 inhibitors UO126 or PD98058 (Calbiochem, San Diego, USA).
Cell proliferation and viability
Cell viability was determined by MTT assay (Sigma-Aldrich, Poole, UK). To determine HFL-1 and PASMC proliferation, cells were grown to 50%–60% confluence on 96-well plates and then incubated in starvation medium (DMEM supplemented with 0% FCS) for 16 h. Cells were then washed and cultured for a further 24 h in starvation medium, full growth medium (10% FCS), MCP-1 (R&D Systems, Abingdon, UK) and chemokine CC receptor-2 (CCR2) antagonist (Calbiochem, San Diego, USA) or in supernatants from strained or unstrained cells. Supernatants were aspirated after a further 24 h of culture, cells frozen at −80°C and analysed by the CyQUANT assay (Sigma-Aldrich, Poole, UK) as per the manufacturer's protocol. Proliferation was expressed relative to unstimulated controls (starvation medium, given a value of 1).
MCP-1 ELISA
Twenty-four hours after CMS, media were collected and cleared by centrifugation (10 000 ×g for 1 min). MCP-1 concentration was measured by ELISA according to the manufacturer's instruction (R&D Systems, Abingdon, UK).
Western blotting
Cells were scraped from the BioFlex membrane and homogenised in cell lysis buffer (Cell Signalling, New England Biolabs, Hitchin, UK). Whole rat lungs were snap frozen in liquid nitrogen and stored at −80°C. Frozen lung (approximately 100 mg) was homogenised in ice cold cell lysis buffer (Cell Signalling, New England Biolabs, Hitchin, UK) containing protease inhibitors. Western blotting was carried out according to standard techniques described in the online supplement.
RNA extraction and real-time qPCR
Total RNA (1 μg) was transcribed to cDNA using L-AMV reverse transcriptase (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. Real-time quantitative PCR was performed using SYBR green master mix (Qiagen, Manchester, UK) or on a Corbett Rotor-Gene 6000 (Qiagen, Manchester, UK). The relative expression of target genes was quantified using the ΔΔCt method normalised to the average expression of two housekeeping genes (HLMEVC and Ea.Hy 926: TUBB and GUSB, HFL-1 and PASMC: GUSB and PPIB, and whole rat lung: ARBP and HRPT). Identification of the optimal type and number of housekeeping genes were determined using GeNorm.27 Primers for α smooth muscle actin (αSMA) were purchased from Qiagen (Manchester, UK) and the remainder was purchased from Invitrogen (Carlsbad, USA).
In vivo MI model of HF
All experiments were carried out in accordance with the United Kingdom Home Office Animals (Scientific Procedures) Act 1986, which conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996). The post-MI HF model in male Sprague–Dawley rats, in which the left coronary artery (LCA) is ligated through a thoracotomy, has been previously described.24–26 ,28 The lung and blood samples of the rats used in these experiments were sourced from previously reported studies.25 ,28
From an initial cohort of animals at 16 weeks post-MI, LVEDP was recorded via apical cardiac puncture with a 2-F Millar catheter (n=6), as previously described.25 ,28 Age-matched control (AMC) animals previously showed no haemodynamic difference to sham-operated animals and those treated with null virus and so they were used as controls.25 ,28 Blood was collected by direct aspiration from the inferior vena cava, centrifuged at 15 000 rcf and stored at −20°C. Serum samples were available for 11 AMC and 16 post-MI animals.
Lungs were removed en bloc and wet weight recorded. Right lungs were snap frozen in liquid nitrogen for subsequent extraction of mRNA (AMC=7, MI=9) or homogenised, weighed and freeze dried for 24 h giving a wet:dry weight (n=10). Left lungs were lavaged with ice cold normal saline (10 mL: bronchoalveolar lavage (BAL)). In a proportion of animals, lungs were excluded owing to macroscopic damage at the time of euthanasia, with the result that BAL was collected from eight AMC and 11 MI rodents.
In a second cohort of animals (n=5), in addition to HF and controls, lungs and serum were harvested from rats with HF infected with AAV9.SERCA2a (2×1011 drp) as previously reported.25 Briefly, animals received gene therapy via tail vein injection 14–16 weeks post-MI and were sacrificed after a further 4–6 weeks. To investigate pulmonary remodelling, whole lungs from animals (AMC=6, MI=6 and MI+SERCA=7) were immediately snap frozen in liquid nitrogen and stored at −80°C for the measurement of lung collagen.
Total lung collagen content measurement
Total lung collagen was determined by measuring hydroxyproline content as previously described.29 Briefly, free hydroxyproline was generated from acid hydrolysis of pulverised lung tissues and derivatised with 4-chlor-7-nitrobenzo-2-oxa-1, 3-diazole to generate a hydroxyproline derivative, which is then analysed with reverse phase high-performance liquid chromatography (HPLC) with fluorescence detection.
Statistical analyses
Data from cellular experiments and in vivo experiments where n≤10 were analysed using Mann–Whitney U test for two groups or Kruskal–Wallis with Dunn's post-test correction for three or more data sets. Data from in vivo experiments with n≥10 were analysed using Student t test if the data were normally distributed.
Results
Cyclic mechanical strain enhanced MCP-1 protein and gene expression by HLMVEC and Ea.Hy 926 cells in an ERK-MAPK-dependent manner
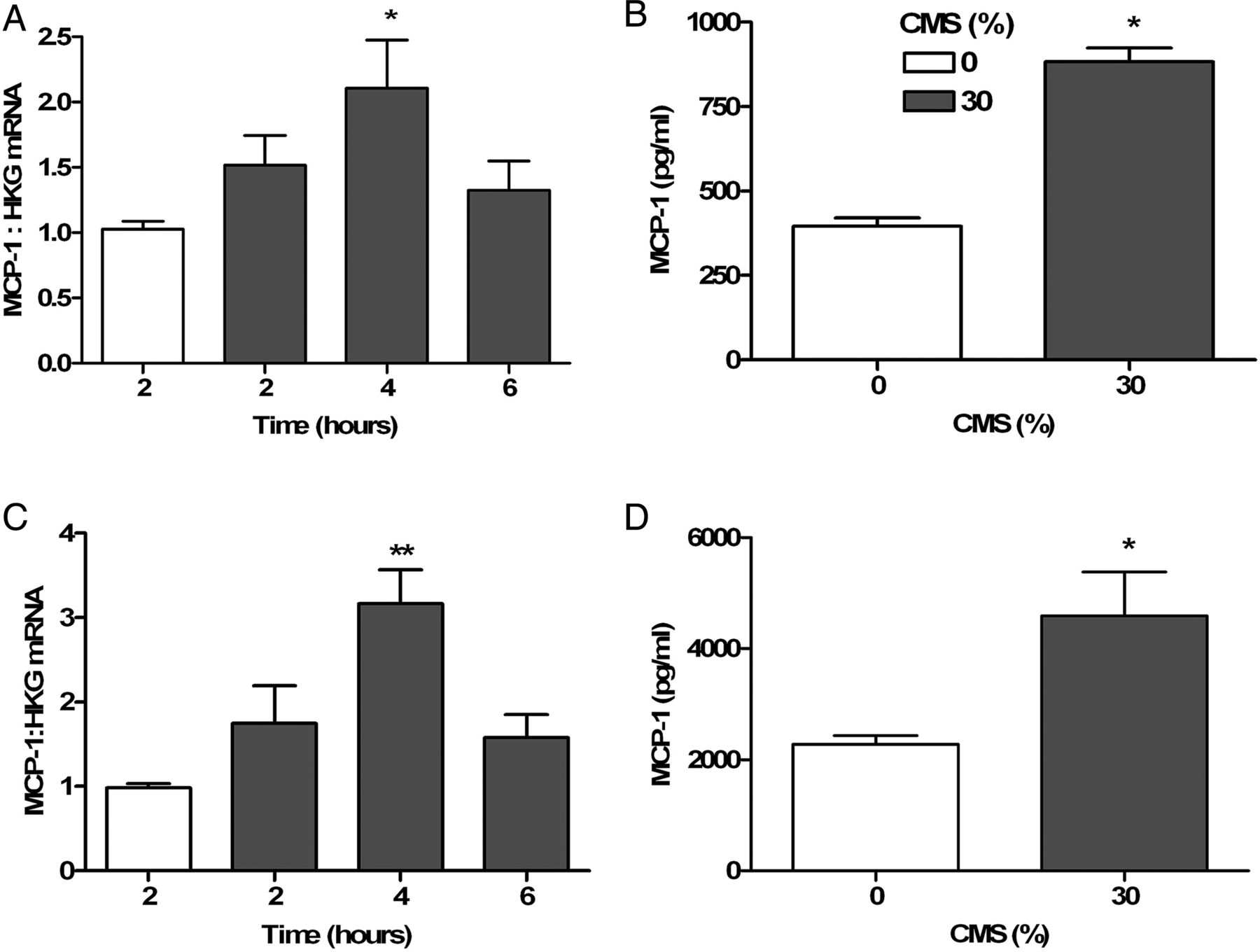
HLMVEC and Ea.Hy 926 cells subjected to CMS released MCP-1 protein (figure 1B, D) and increased MCP-1 gene expression peaking at 4 h (figure 1A, C: HLMVEC: 1.028±0.146 (0 h) vs 2.108±0.909 (4 h); n=4; Ea.Hy 926: 0.981±0.10 (0 h) vs 3.17±0.80 (4 h), n=4, p<0.05 Mann–Whitney). CMS induced maximal p42/44 extracellular-signal-regulated kinase (ERK) phosphorylation at 30 min (figure 2A, B). Pharmacological inhibition of the ERK pathway using UO126 and PD95069 (both 3 µM) attenuated CMS-induced MCP-1 gene expression in and protein release by Ea.Hy 926 cells (figure 2C, D). Inhibition of JNK and p38 MAPK pathways did not alter MCP-1 expression (data not shown).

Cyclic mechanical strain (CMS; 30%; 0.33 Hz) increased monocyte chemoattractant protein-1 (MCP-1) gene and protein expression by human lung microvascular endothelial cells (HLMVEC) and Ea.Hy 921 cells. (A) (HLMVEC) and (C) (Ea.Hy 921 cell): time course of mRNA expression in response to CMS (shaded bars) compared with unstretched controls (open bars); (B) (HLMVEC) and (D) (Ea.Hy 921): MCP-1 concentration in supernatants after CMS (30%, 0.33 Hz, 4 h: shaded bars) and incubation for a further 24 h compared with unstretched controls (open bars). Gene expression experiments (A (n=6) and C (n=4)) analysed using Kruskal–Wallis non-parametric test with Dunn's post-test comparison. Protein expression (B (n=4) and D (n=6)) analysed using Mann–Whitney U test; *p<0.05; **p<0.01.

(A) and (B) cyclic mechanical strain (CMS; 30%; 0.33 Hz) increased phosphorylation of extracellular-signal-regulated kinase (ERK) 42 and ERK 44 at 10 and 30 min by Ea.Hy 926 cells (n=4; Kruskal–Wallis with Dunn's post-test comparison). Pharmacological inhibition of the ERK signalling pathway by UO126 and PD95069 (3 µM) attenuated CMS-induced monocyte chemoattractant protein-1 protein release at 24 h (C; n=6 vehicle and UO126, n=4 PD98059; Kruskal–Wallis with Dunn's post-test comparison) and mRNA expression at 4 h (D; n=5 vehicle and UO126, n=4 PD98059; Kruskal–Wallis with Dunn's post-test comparison).
Conditioned media from cells subjected to cyclic mechanical strain-induced fibroblast and smooth muscle cell proliferation and fibroblast differentiation
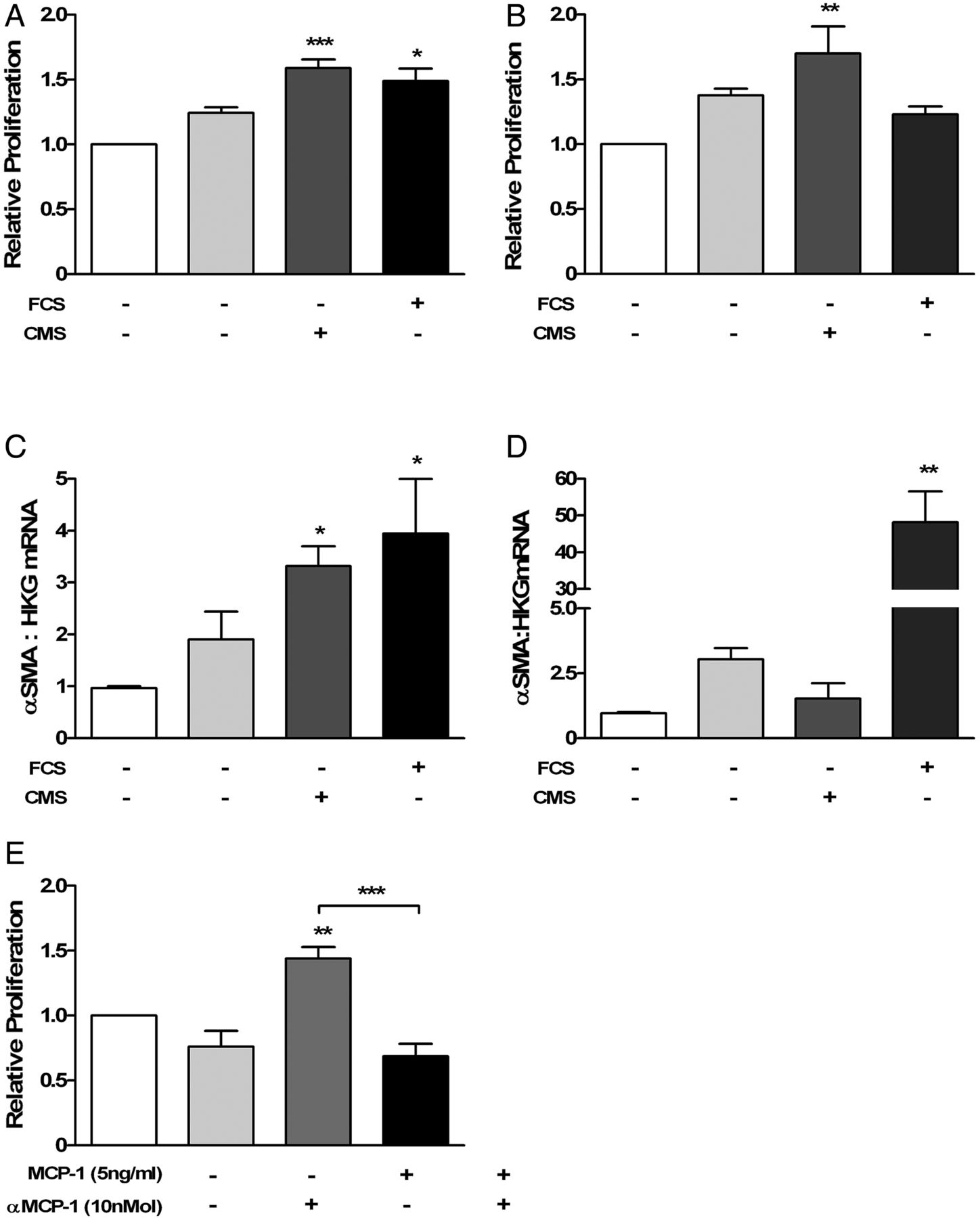
Incubation with conditioned media from EC subjected to CMS (30%) increased HFL-1 and PASMC proliferation (figure 3A, B). αSMA gene expression increased in HFL-1 cells (3.32±0.94, n=5; p<0.05) but not PASMC (1.53±1.3, n=5; Kruskal–Wallis with Dunn's post-test; figure 3C, D). Stimulation with exogenous MCP-1 (0.1–10 ng/mL) increased proliferation of PASMC, with a maximum effect at 5 ng/mL (relative proliferation 1.603±0.08; p<0.001; one-way analysis of variance (ANOVA) with Dunnet's post-test; n=3). CCR2 receptor antagonist (10 nMol) abolished the effect of MCP-1 on MCP-1 (1 ng/mL)-induced proliferation in PASMC (MCP-1 plus CCR2 receptor antagonist (0.69±0.19) compared with MCP-1 alone (1.44±0.18), n=4, p<0.001 one-way ANOVA with Dunnet's post-test: figure 3E). Proliferation of HFL-1 was unaffected by MCP-1 (data not shown).

Supernatants from human lung microvascular endothelial cells exposed to cyclic mechanical strain (CMS; 0 or 30% elongation) increased cellular proliferation of HFL-1 (A; n=6; Kruskal–Wallis with Dunn's post-test). Supernatants from Ea.Hy 926 cells exposed to CMS (0 or 30% elongation) increased cellular proliferation of PASMC (B; n=4) and increased α-smooth muscle actin gene expression by HFL-1 cells (C; n=5) but not by PASMC (D; n=5). CCR2 antagonist-attenuated MCP-1 induced PASMC cellular proliferation (E; p<0.01 cells incubated with MCP-1, 1 ng/mL (1.44±0.18) compared with cells incubated with CCR2 antagonist 10 nM only (0.76±0.24); p<0.001 MCP-1, 1 ng/mL (1.44±0.18) compared with MCP-1 1 ng/mL with CCR2 antagonist 10 nM (0.69±0.19); n=4; one-way analysis of variance with Dunn's post-test). FCS, 10% fetal calf serum; MCP-1, monocyte chemoattractant protein-1; CCR2, chemokine CC receptor-2; PASMC, pulmonary artery smooth muscle cell.
Effect of HF on systemic and pulmonary induction of MCP-1
Having confirmed that CMS stimulated MCP-1 release by vascular endothelium, which had the potential to induce remodelling, we investigated whether this pathway was activated in an animal model of post-MI HF characterised by elevated LVEDP and PVH. Compared with AMCs, rats with post-MI HF had increased LVEDP, heart weight:body weight (HW:BW) and lung weight:body weight (LW:BW) ratios without an increase in wet:dry lung weight (table 1). Serum MCP-1 levels were elevated in samples from rats with HF (figure 4A; 102.10±33.05 vs 72.51±19.81 pg/mL, HF (n=16) vs AMC (n=11): p=0.014) and this was more marked in animals with the largest infarcts (HW:BW ratio >4 and full thickness infarct in the left anterior descending coronary artery territory involving the LV apex30: 110.3±31.8, n=10; p=0.013; Student t test). BAL fluid levels of MCP-1 were not elevated in all rats with HF, but were in those with large infarcts (figure 4B). MCP-1 and IL-6 gene expression in whole lung from rats with HF were elevated compared with the AMC group (figure 4C).
Parameters from HF rodents at 16 weeks postmyocardial infarction or AMC

Serum monocyte chemoattractant protein-1 (MCP-1) concentrations were elevated in all rodents with heart failure (HF), a finding that was more pronounced in animals with a large myocardial infarct (HF (L)); A; aged-matched control (AMC) n=11, HF (S/M) n=6, HF (L) n=10). Bronchoalveolar lavage (BAL) levels of MCP-1 were not higher in all rodents with HF, but were in those that had a large HF (HF (L)); B: BAL: AMC n=8, HF (S/M) n=4, HF (L) n=7). (C) Gene expression of MCP-1 and interleukin-6 were elevated in whole lungs from rodents with HF (AMC n=7, HF n=9: GOI, gene of interest; HKG, housekeeping genes). Student unpaired t test was used to compare groups; *p<0.05, **p<0.01, ***p<0.001, #p=0.76.
Increased pulmonary MCP-1 gene expression and early structural remodelling was attenuated after rescue of the HF phenotype with SERCA2a gene therapy
Rats with HF treated with SERCA2a gene therapy had increased HW:BW ratios, but not increased LW:BW, LVEDP and PVH at 4–6 weeks after gene delivery, consistent with the lungs having been rescued from the haemodynamic effects of HF (table 2). SERCA2a mRNA and protein were not detected in the lungs of rodents from all three groups by PCR and western blotting, respectively (see online supplementary figure S1), confirming this was not a direct effect of SERCA2a delivery to the pulmonary vasculature. Neither serum nor BAL MCP-1 levels were significantly altered in rats with post-MI HF after SERCA2a gene therapy (figure 5A, B); however, SERCA2a gene therapy reduced MCP-1 and IL-6 gene expression in rats with post-MI HF compared with controls (figure 5C, D). Whole lung collagen levels, an indicator of structural remodelling, were elevated in animals with post-MI HF, but not in animals treated with SERCA2a gene therapy (figure 5E).
Parameters from HF rodents at 20–22 weeks postmyocardial infarction±SERCA2a gene therapy and AMCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
There was no difference in monocyte chemoattractant protein-1 (MCP-1) concentrations in rat serum (A; n=5) and bronchoalveolar lavage (B, n=5) after myocardial infarction (heart failure, HF) or in animals treated with SERCA gene therapy (HF+SERCA). Interleukin-6 gene expression levels were elevated in whole lungs of animals postmyocardial infarction HF compared with age-matched control animals (AMC: D, n=5), MCP-1 gene expression levels were reduced in SERCA-treated compared with HF-alone animals (C, n=5). Collagen content of lungs measured by high-performance liquid chromatography was increased in rodents with HF (E: HF; n=6 vs AMC n=6) and attenuated after treatment with gene therapy (HF+SERCA, n=7). Kruskal–Wallis with Dunn's post-test comparison was used throughout; *p<0.05, **p<0.01.
Discussion
HF is associated with significant dyspnoea that limits quality of life and is due, at least in part, to structural remodelling of the alveolar capillary membrane. MCP-1 is one of several mediators of inflammation and remodelling that are elevated in HF, and circulating levels of MCP-1 from patients with HF correlate with disease severity and prognosis.13 The source of mediators in HF and the mechanisms underpinning structural remodelling of the alveolar capillary membrane are unclear. Here, we have demonstrated that MCP-1 gene expression was induced in human EC subjected to CMS in vitro, in an ERK 1/2 dependent manner. Supernatants from EC subjected to CMS stimulated proliferation of human mesenchymal cells and α-SMA expression in a fetal fibroblast cell line (HFL-1) but not in PASMC. Similarly, PASMC but not HFL-1 cells proliferated in response to MCP-1. Finally, in a rodent post-MI model of HF, larger myocardial infarcts were associated with an increased concentration of MCP-1 in serum and BAL. After SERCA2a gene therapy, which attenuated the increase in PVH after MI, the induction of MCP-1 and the total lung collagen content were reduced.
We hypothesised that PVH and resultant mechanical strain at the pulmonary microvasculature contributes to structural remodelling. Consistent with this, HLMVEC and Ea.Hy926 cells upregulated MCP-1 gene expression and protein release in response to CMS in an ERK-dependent manner (figures 1 and 2). Other ECs are similarly mechanosensitive, upregulating MCP-1 when subjected to CMS.21 ECs from different tissue beds respond differentially to CMS and this is the first report of upregulation of MCP-1 in response to CMS in HLMVEC cells. The role for the ERK signalling pathway in EC mechanotransduction is not unheralded: CMS-induced MCP-1 release by EC involved the small g-protein Rac and reactive oxygen species, with a subsequent increase in AP-1-dependent gene expression.21 ,31
Conditioned media from EC subjected to CSM increased PASMC and fibroblast proliferation and fibroblast differentiation (αSMA expression: figure 3A–D), suggesting that PVH-associated mechanical strain could provide a stimulus for pulmonary structural remodelling. Because the effect of stretch-induced proliferation was small (comparing the effects of media from stretched and unstretched EC) in these short-term experiments, it was not possible to conclusively demonstrate dependence on MCP-1 using a CCR2 antagonist. However, exogenous MCP-1 stimulated proliferation that was abolished using the CCR2 antagonist (figure 3E), suggesting a potential role for MCP-1 in remodelling. MCP-1 is a potent mononuclear cell chemoattractant that is derived from several cell types. It is overexpressed in the lungs of patients with fibrotic lung disease,32 and circulating levels are elevated in patients with HF.33 Traditional roles ascribed to MCP-1 in the induction of lung fibrosis have included recruitment of M2-activated macrophages and fibrocytes,34 and stimulation of fibroblasts to produce procollagen and transforming growth factor β.35 The lack of response of HFL-1 to MCP-1 stimulation has many possible interpretations. For example, HFL-1 may not be the most relevant cell type to model lung remodelling, MCP-1 may be acting through other cell types such as vascular smooth muscle or through mechanisms other than proliferation and the fibroblast proliferation induced by MCP-1 may be indirect requiring prostaglandin E2 production by epithelial cells as has been found by others.36
The lungs of rats with HF were heavier, but not wetter, than those from control animals, suggesting structural remodelling, supported by elevated collagen levels in the lungs from rats with HF after MI (table 1 and figure 5E). Preliminary histological assessment of the lungs of these rats demonstrated that the collagen deposition was subtly increased compared with untreated controls. Therefore, measurement of lung collagen by HPLC was used because it provides a more sensitive and quantitative assessment of lung collagen.30 SERCA2a gene therapy attenuated the increase in LVEDP after MI and provides a method for investigating the effect of reducing PVH while not affecting the size of the myocardial infarct.37 ,38 In addition to showing improved cardiac haemodynamics, SERCA2a gene therapy animals did not have an increased lung weight and had lower collagen levels, suggesting a reduction in, and possibly even a reversal of, early structural remodelling (table 2 and figure 5E). Furthermore, the decrease in HF-related MCP-1 and IL-6 expression after SERCA2a therapy implies that reducing PVH may have contributed to attenuating the stimulus for mediator release. The failure to detect SERCA2a expression in whole lungs extracts by either PCR or western blotting addressed the possibility that SERCA2a affected the lung directly after systemic delivery (see online supplementary figure S1). Furthermore, while AAV vectors have tropism for vascular smooth muscle, it is the SERCA2b isoform that is expressed in that tissue.39
The alteration of gene expression in the lungs in response to SERCA2a gene therapy is a novel finding, which may have contributed to the beneficial effects of gene therapy in similar animal models38 that have encouraged translation into clinical studies.40 Indeed, symptom burden and exercise limitation were reduced in patients with HF receiving SERCA2a gene therapy in the recently reported phase2a Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease trial.41 Reversal of PVH in the lungs may contribute to these effects and, by removing the driving stimuli for both inflammatory mediator release and structural remodelling, may improve long-term outcomes and symptom control. Although potentially beneficial, the reduction in collagen associated with SERCA2a gene therapy must be approached with caution. If reversal of pulmonary remodelling outpaces improvement in cardiac haemodynamics, then the protection that structural remodelling provides against cardiogenic pulmonary oedema may be lost. Conversely, the potential to reverse remodelling in pulmonary lymphatic vessels subjected to increased flow if the same mechanisms applied would tend to protect against pulmonary oedema.42 Consequently, understanding the effects of gene therapy on both the lung and the heart in the clinical setting will be vital to the overall success of the intervention.
The in vitro stretch and, to a lesser extent, the rodent models used in this work suffer from the inevitable deficiencies of short-term models of chronic and complex clinical conditions. In the complex post-MI state, MCP-1 alone is unlikely to be solely responsible for lung remodelling; we have also identified elevated IL-6 levels (figure 4). Similarly, work by others has addressed other mediators, such as endothelin-1, although outcomes in both clinical trials of endothelin antagonists in HF and animal models have been negative.43 ,44 In addition, our modelling of the mechanical effects of PVH on the microvascular endothelium using CMS via the Flexercell system is reductionalist, but has the benefit of using HLMVECs, potential upstream targets of elevated LAP. Similarly, the LCA ligation model of HF has inherent limitations. We have compared AMC animals rather than sham-operated animals; however, preliminary data did not show differences in lung, heart and body weights nor MCP-1 levels between these groups (see online supplementary table S1). The benefit of studying SERCA gene therapy is to provide an assessment of the effect of attenuated LAP elevation without affecting the infarct itself. Future experiments, outside the scope of this work, should use animals that do not express MCP-1. By suggesting a role for MCP-1, we do provide a potential therapeutic target for future investigation, which has also been implicated in structural and vascular remodelling in other pulmonary pathologies, including pulmonary hypertension.45
In conclusion, CMS induced MCP-1 expression in HLMVEC in an ERK-dependent manner. Conditioned medium from EC exposed to CMS stimulated fibroblast differentiation, PASMC and fibroblast proliferation, both features of pulmonary structural remodelling. SERCA2a gene therapy in a post-MI HF model not only rescued cardiac function and reduced LVEDP, but also attenuated pulmonary MCP-1 and IL-6 gene expression, and total lung collagen levels a marker of structural remodelling. These findings suggest a mechanism underlying pulmonary remodelling in HF, whereby PVH induces lung microvascular ECs to produce proinflammatory and profibrotic mediators like MCP-1. These findings point to novel, potential therapeutic targets for the relief of dyspnoea in patients with HF.
Acknowledgments
SERCA Gene Therapy was kindly gifted by Prof R. Hajjar, Cardiovascular Research Centre, Mount Sinai School of Medicine, New York, NY, USA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors JESP wrote the manuscript and undertook all research experiments except for rodent surgical procedures and whole lung collagen measurement. ARL performed the rodent surgical procedures and haemodynamic measurements, and edited the manuscript. LRH assisted with the rodent experiments. DS performed additional experiments (MCP-1-stimulated proliferation) after review. HX performed the whole lung collagen measurement. PO assisted with rodent experiments. LP assisted with mechanical stretch experiments. RCC oversaw the whole lung collagen experiments. SJW co-supervised this work and edited the manuscript. MJDG supervised this research and edited the manuscript.
Funding British Heart Foundation (FS/11/67/2894), Medical Research Council (G0500373, G0701361).
Competing interests JESP was supported with a Clinical Training Fellowship from the Medical Research Council to carry out this work. This work was also supported by the National Institute of Health Research (NIHR) funded Cardiovascular and Respiratory Biomedical Research Units of the Royal Brompton and Harefield NHS Foundation Trusts.
Provenance and peer review Not commissioned; externally peer reviewed.