Article Text

Abstract
Background COPD is a complex chronic disease with poorly understood pathogenesis. Integrative genomic approaches have the potential to elucidate the biological networks underlying COPD and lung function. We recently combined genome-wide genotyping and gene expression in 1111 human lung specimens to map expression quantitative trait loci (eQTL).
Objective To determine causal associations between COPD and lung function-associated single nucleotide polymorphisms (SNPs) and lung tissue gene expression changes in our lung eQTL dataset.
Methods We evaluated causality between SNPs and gene expression for three COPD phenotypes: FEV1% predicted, FEV1/FVC and COPD as a categorical variable. Different models were assessed in the three cohorts independently and in a meta-analysis. SNPs associated with a COPD phenotype and gene expression were subjected to causal pathway modelling and manual curation. In silico analyses evaluated functional enrichment of biological pathways among newly identified causal genes. Biologically relevant causal genes were validated in two separate gene expression datasets of lung tissues and bronchial airway brushings.
Results High reliability causal relations were found in SNP–mRNA–phenotype triplets for FEV1% predicted (n=169) and FEV1/FVC (n=80). Several genes of potential biological relevance for COPD were revealed. eQTL-SNPs upregulating cystatin C (CST3) and CD22 were associated with worse lung function. Signalling pathways enriched with causal genes included xenobiotic metabolism, apoptosis, protease–antiprotease and oxidant–antioxidant balance.
Conclusions By using integrative genomics and analysing the relationships of COPD phenotypes with SNPs and gene expression in lung tissue, we identified CST3 and CD22 as potential causal genes for airflow obstruction. This study also augmented the understanding of previously described COPD pathways.
- COPD Pathology
- Emphysema
Statistics from Altmetric.com
Key messages
What is the key question?
-
What are the causal genetic variants changing gene expression in the lung that in turn associate with lower lung function and COPD?
What is the bottom line?
-
Lung function-associated genetic variants alter the mRNA expression of nearby genes involved in biological pathways underpinning pulmonary function and COPD pathogenesis.
Why read on?
-
New genes of airflow obstruction are identified with a generalised framework for the identification of causal genes from joint examination of genome-wide genotyping and gene expression data in the same patients.
Introduction
Genome-wide association studies (GWAS) have revolutionised our ability to identify common genetic variants that are associated with complex chronic diseases.1 This approach has been applied to COPD, a lung disease that is caused predominantly by cigarette smoking in the western world.2–4 It is well known that only a subset of heavy smokers (15–20%) develop clinically relevant COPD and there is considerable evidence that there is a substantial genetic component involved in its pathogenesis.5 Although GWAS have identified novel loci that harbour susceptibility genes, they do not allow precise identification of the causal variant (or variants). In addition, GWAS do not provide information on how and to what extent the gene (or genes) within the susceptibility loci contribute to the phenotype. Interestingly, the majority of genetic variants which have been associated with disease traits by GWAS do not affect the coding sequence of genes but are located in intergenic regions or introns.6 Possible explanations are that the associated alleles are in linkage disequilibrium (LD) with rarer coding alleles with large effect sizes7 and/or that the genetic variants control the level of expression of genes involved in pathogenetic pathways. For complex genetic diseases, such as COPD, the effects of susceptibility alleles may primarily act by regulating gene expression rather than by altering protein coding as in most Mendelian diseases.8
We recently reported the discovery of a large number of lung-specific expression quantitative trait loci (eQTLs)9 and identified the most likely causal genes within three GWAS-nominated COPD susceptibility loci.10 The aim of the present study is to use the power of genome-wide mRNA expression arrays combined with genome-wide interrogation of single nucleotide polymorphisms (SNPs) to pinpoint specific SNPs that are related to lung tissue gene expression and to COPD phenotypes. Identification of susceptibility alleles that function as strong eQTLs increases the likelihood of identifying the true susceptibility gene within loci with large areas of LD.8 Moreover, the use of integrative genomics by combining environmental exposure data with susceptibility alleles, RNA expression levels, and different COPD phenotypes can unravel causal genetic relationships.11 The basic principle of the current study is to map the genetic regulation of gene expression to identify DNA variants that induce changes in transcriptional networks that in turn contribute to COPD and airway obstruction pathogenesis.
Methods
Subject selection
The methods for subject selection and phenotyping, and for interrogation of gene expression and genotype were recently described.9 The lung tissue used for discovery of eQTLs was from 1111 human subjects who underwent lung surgery at three academic sites, Laval University, University of British Columbia (UBC) and University of Groningen, henceforth referred to as Laval, UBC and Groningen, respectively. All lung specimens from Laval were obtained from patients undergoing lung cancer surgery and were harvested from a site distant from the tumour. At UBC, the majority of samples were from patients undergoing resection of small peripheral lung lesions. Additional samples were from autopsy and at the time of lung transplantation. At Groningen, the lung specimens were obtained at surgery from patients with various lung diseases, including patients undergoing therapeutic resection for lung tumours, harvested from a site distant from the tumour, and lung transplantation. For the present study, the principal aim was to examine smoking-related airway obstruction. Thus, we excluded subjects whose lung function may have been influenced by lung diseases other than COPD and lung cancer. Exclusion criteria are provided in the online supplement.
COPD phenotypes and GWAS
Genome-wide association was performed using linear or logistic regression models on the three phenotypes: FEV1% predicted and FEV1/FVC as continuous variables, and COPD defined dichotomously based on an FEV1/FVC<0.7 cut-off (see online supplement). Single-marker association tests were run within each cohort adjusting for age, gender and smoking status. Fixed-effects meta-analysis was then performed combining the three cohorts using inverse SE weighting.
Expression trait processing
Expression traits were adjusted for age, gender and smoking status as described previously.9 Gene expression data are available in the Gene Expression Omnibus repository through accession number GSE23546.
Causality models
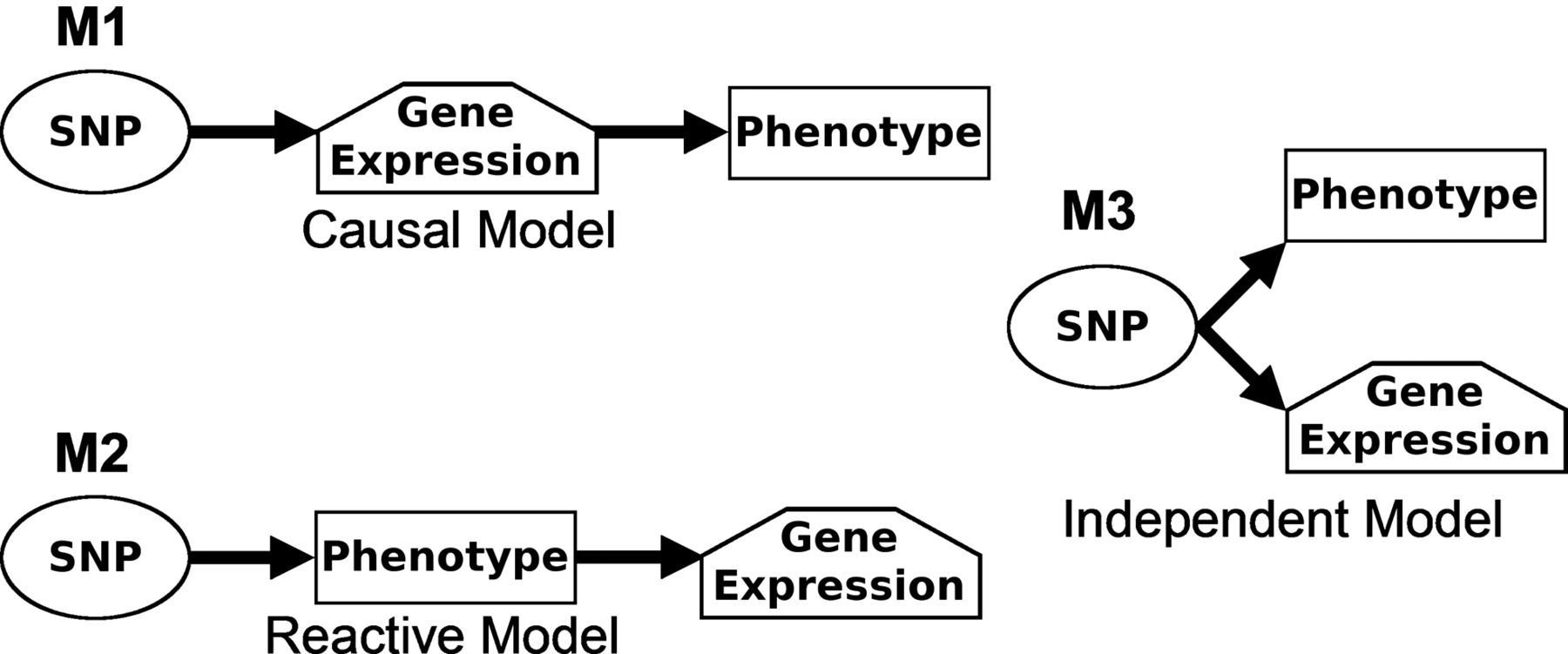
We evaluated three competing causality models to describe the relationships between lung eQTL-SNPs, RNA expression and COPD phenotypes12 (figure 1). Model 1 indicates a potential causal relationship where the SNP acts on gene expression to produce the phenotype, which was the main interest of this work. Model 2 indicates that the gene expression pattern is reactive to the phenotype, that is, the phenotype drives gene expression. Model 3 is the independent model where the SNP acts on the phenotype and gene expression independently. We required a p value <1×10−3 for SNP associated with phenotype before examining a particular variable triplet (ie, SNP, gene expression and phenotypes). The causal model (model 1) was selected when p value <0.05 was found for SNP associated with gene expression adjusting for phenotype and p value >0.05 was found for SNP associated with phenotype adjusting for gene expression. More details are provided in the online supplement. Analyses were performed in the three cohorts separately and then combined into a meta-analysis. We then conducted sample bootstrapping and repeated the causality test for N=1000 realisations. The reliability score of the molecular relationship is the fraction of bootstrap realisations that supports the call observed. Threshold for p values, number of bootstrap reselections and reliability cut-off (>0.8) were selected based on previous literature.12

Causality models showing the relationships between the expression of a gene, a phenotype and a single nucleotide polymorphism (SNP). Three models are depicted including a causal model (M1), a reactive model (M2), and an independent model (M3). M1 is the simplest, and states that the genotype at an expression quantitative trait loci SNP acts directly on gene expression pattern to produce the phenotype. M2 states that the gene expression pattern is reactive to the phenotype, and M3 states that the gene expression pattern and phenotype are independent.
Manual curation and pathway analyses on reliable causal models
The biology and possible role of genes in the causal model were reviewed by manual curation and bioinformatics tools, including Ingenuity Pathway Analysis (IPA), MetaCore and Partek (see online data supplement).
Replication studies
Biologically relevant causal genes were validated in two datasets. First, a bronchial airway epithelial dataset where genome-wide gene expression levels were obtained from bronchial brushing of 238 individuals and associated with lung function as previously described.13 Second, a regional lung tissue dataset where genome-wide gene expression was obtained for eight regions of the same lung and for eight patients. The association between gene expression levels and micro-CT-based regional emphysema severity was assessed.14 More details are provided in the online data supplement.
Results
Of the 1111 subjects in whom there were data on genotype and gene expression, 848 with sufficient phenotypic information were included in the analysis. The demographic and clinical features of the subjects in the three cohorts are described in table 1.
Clinical characteristics of patients
Causal pathways that fit model 1
Model fitting was restricted to SNPs that were significantly associated with at least one of the lung function variables or COPD as a categorical variable at a p value cut-off of 10−3. SNPs were considered an eQTL if they had a p value ≤10−5. Using these criteria, there were 4465 SNP–mRNA–phenotype triplets. Of these, 249 triplets showed a significant fit to the causal model for at least one of the phenotypes with a reliability score >0.8 in at least one cohort and/or in the meta-analysis. A fit to this model means that the SNP was associated with one of the phenotypes and affected gene expression in a direction that supported a causal relationship.
The list of causal models with a confidence score >0.8 in any one of the cohorts or in the meta-analysis is shown in online supplementary table S1. For FEV1% predicted, 169 causal models were found, including 122 unique SNPs and 169 probe sets. The 169 models included 168 cis eQTLs and 1 trans eQTL, respectively. For FEV1/FVC, 80 causal models were found involving 63 unique SNPs and 80 probe sets. Among these 80 models, 79 were cis eQTLs and 1 was a trans eQTL. No causal model was found when using COPD as a categorical variable. There was no overlap between causal pathway models for FEV1% predicted and FEV1/FVC.
Manual curation of significant and reliable causal models
Significant causal models were inspected manually. We identified a number of models of potential biological relevance for FEV1% predicted and FEV1/FVC. The most biologically relevant models are listed in table 2. The p values and direction of effect for each of the SNP associations with the gene expression and the phenotype are also indicated. For example, SNP rs6048956 was significantly associated with cystatin C (CST3) transcript. The common allele was associated with higher expression of the transcript (figure 2 and positive eQTL Z score in table 2) and with lower FEV1% predicted (figure 2 and negative Z score with phenotype in table 2). Taken together, these results suggest that the common allele confers susceptibility to a lower FEV1% predicted value through upregulation of the CST3 mRNA expression levels in the lung. This was confirmed in a second causality model that interrogated a different probe set for CST3, FEV1% predicted and rs6515375 (table 2 and see online supplementary figure S1). Analogous observations were made for natural cytotoxicity triggering receptor 3 (NCR3), cystatin A (CSTA), peroxisome proliferation-activated receptor γ coactivator 1α (PPARGC1A), folliculin (FLCN), BCL2-like 1 (BCL2L1), cell adhesion molecule 2 (CADM2), and tumour necrosis factor receptor superfamily member 10b (TNFRSF10B) for FEV1% predicted, and glutathione S-transferase ω 2 (GSTO2), DEP domain containing 6 (DEPDC6), CD22 molecule (CD22), mucin 22 (MUC22), and serine peptidase inhibitor Kazal type 5 (SPINK5) for FEV1/FVC (table 2). The direction of effects for all these causality models are illustrated in online supplementary figures S2–13.
Models of biological relevance identified by manual curation

{kind=link}
{kind=link}
Direction of effects for causality model with rs6048956, mRNA expression of cystatin C (CST3) and FEV1% predicted. The left, centre and right panels show the results for Laval, University of British Columbia (UBC) and Groningen samples, respectively. (A–C) are boxplots of gene expression levels in the lung for CST3 according to genotype groups for single nucleotide polymorphism (SNP) rs6048956. The left y-axis shows the mRNA expression levels for CST3. The x-axis represents the three genotype groups for SNP rs6048956. The right y-axis shows the proportion of the gene expression variance explained by the SNP (black bar). Box boundaries, whiskers and centre mark in boxplots represent the first and third quartiles, the most extreme data point which is no more than 1.5 times the IQR, and median, respectively. (D–F) are barplots of the mean and SE of FEV1% predicted according to genotype groups for SNP rs6048956.
Pathway analyses
To find key biological pathways involved in COPD, causality genes were overlaid on canonical pathways available in IPA. One hundred and sixty-nine genes and 80 causality genes for FEV1 and FEV1/FVC were considered, respectively (see online supplementary table S1). Table 3 shows all canonical pathways enriched with causality genes (p<0.05). Of interest was the aryl hydrocarbon receptor signalling pathway, which is involved in xenobiotic clearance. Five causality genes were noted in this canonical pathway, including ARNT (aryl hydrocarbon receptor nuclear translocator), IL6 (interleukin 6), GSTO2, ALDH8A1 (aldehyde dehydrogenase 8 family, member A1) and TRIP11 (thyroid hormone receptor interactor 11). The aryl hydrocarbon receptor signalling pathway and the location of the causality genes are illustrated in online supplementary figure S14. Another pathway involved in xenobiotic handling, the xenobiotic metabolism signalling pathway, was also enriched for causality genes, many of which overlapped with those in the aryl hydrocarbon receptor signalling pathway (ARNT, IL6, GSTO2 and ALDH8A1). The former also includes PPARGC1A and PRKCE (protein kinase C, ε). This pathway is illustrated in online supplementary figure S15.
Canonical pathways enriched for causality genes using the Ingenuity Pathway Analysis system
For the Partek analyses 118 and 45 transcripts were identified for FEV1% predicted and FEV1/FVC, respectively. These transcripts mapped to a total of 115 genes in Partek GS (based on official gene symbols) and were parsed to Partek Pathway Suite. Causality genes were overlaid onto canonical pathways from the REACTOME and KEGG databases. Table 4 shows the canonical pathways enriched with causality genes (p<0.05). TNF-related apoptosis-inducing ligand (TRAIL) signalling was the most significant pathway, with several other apoptosis-related pathways included in the list of top significant associations. The glutathione metabolism pathway connects to both the cyanoamino acid and taurine and hypotaurine metabolism pathways (see online supplementary figure S16). These three pathways were identified in our enrichment analysis and were also identified using the IPA system. GO functional categories also showed enrichment for causality genes (see online supplementary figure S17) including the γ-glutamyl transferase activity.
Canonical pathways enriched for causality genes using Partek
Replication of the most biologically relevant causality models
No other large-scale lung eQTL dataset is available in patients with and without COPD. To replicate the most biologically relevant causality models, we relied on two genome-wide expression datasets. Causality genes were first validated in a gene expression study of bronchial airway epithelial cells obtained by bronchoscopy. All 13 genes represented in table 2, except MUC22, were assayed in this airway dataset. Two genes were significantly associated with lung function at a false discovery rate (FDR) of 5% in the bronchial airway epithelial dataset: CSTA (FDR=0.002 with FEV1% predicted) and TNFRSF10B (FDR=9.35×10−5 with FEV1% predicted). In both cases, the direction of effect was the same as that in the current study (table 5). For example, higher CSTA mRNA levels were associated with worse lung function (table 2 and see online supplementary figure S3).
Replication of causality genes in the bronchial airway epithelial and the regional lung tissue datasets
Causality genes identified in this study were also compared with a second lung transcriptomic study that evaluated the impact of regional emphysema severity on gene expression. Interestingly, CD22 was positively associated with regional emphysema severity within individuals (p=5.8×10−5) and was a part of the 127 gene signature for emphysema identified in that study.14 This observation is consistent with the current study showing that carriers of the rare allele for rs10411704 had greater mRNA expression of CD22 and worse FEV1/FVC (table 2 and see online supplementary figure S11). Associations with regional emphysema severity were also observed for three other genes in table 2 including NCR3 (p=0.058), PPARGC1A (p=0.081) and BCL2L1 (p=0.051), but these were not statistically significant. However, the direction of effect was consistent only for PPARGC1A. The expression of this gene was found to decrease with emphysema severity in the regional lung tissue dataset and, in the current study, carriers of the rare allele for rs4550905 were associated with less mRNA expression of PPARGC1A in the lung and lower FEV1 (table 2 and see online supplementary figure S4).
Discussion
This investigation integrated a genome-wide eQTL study on lung tissue with genome-wide genetic association results for lung function and COPD in the same subjects. For the discovery of eQTLs we used the entire dataset which consisted of 1111 tissue samples from subjects who had lung surgery for a variety of reasons. In order to focus on COPD, we limited the study to 848 subjects with sufficient phenotypic information and without a lung disease (other than COPD and lung cancer) which could cause abnormalities of pulmonary function. We limited the causal pathway analysis to SNPs that were both significantly related to gene expression (p<10−5) and were associated with one of three COPD phenotypes (p<10−3), that is, FEV1% predicted, FEV1/FVC and COPD defined as FEV1/FVC<0.7. Of the 4465 SNP–mRNA–phenotype triplets which met our inclusion criteria, 249 triplets showed a significant fit to the causality model for at least one of the phenotypes with a reliability score ≥0.8. We thus provide evidence that these SNPs influence disease susceptibility by altering gene expression. Causality pathway genes were enriched in pathways involved in xenobiotic handling, antiprotease and antioxidant activity and apoptosis.
Two of the eQTL-SNPs in CST3 (rs6515375 and rs6048956) and another for CSTA (rs2270859) were in the causal pathway for FEV1% predicted. The two CST3 SNPs were in perfect LD and were eQTLs for two different probe sets. CST3 and CSTA are cysteine antiproteases; CST3 antagonises cysteine cathepsins such as cathepsin L and S, and CSTA acts similarly for cathepsins B, H and L.15 ,16 Interestingly the direction of association is such that the alleles that are associated with a higher mRNA level of CST3 and CSTA are associated with lower FEV1/FVC.
The above findings might seem paradoxical since cystatins are antiproteases and if one simply invoked the protease–antiprotease hypothesis, then one might expect that individuals with higher levels to be protected from COPD. One possibility is that upregulation of CST3 mRNA and protein could be the result of a feedback loop stimulated by high protease levels. This would be supported by observations in bronchoalveolar lavage fluid and serum reporting higher cystatin C level in patients with emphysema.17 ,18 Alternatively excessive inhibition of proteases may confer biological effects on a yet to be discovered mechanism which contributes to the pathogenesis of airflow obstruction. Further molecular studies of the involved proteins might shed more light on this. We previously reported SNPs associated with CST3 mRNA and protein levels in alveolar macrophages.19 However, in the latter study, higher CST3 mRNA in alveolar macrophages was associated with higher FEV1. Together, these studies suggest that the cystatin genes are important in the pathogenesis of COPD, however the relationship between CST3 mRNA expression levels and lung function remains to be elucidated in relevant tissues and cell types.
With respect to antioxidant activity, eQTLs from PPARGC1A and GSTO2 were found. PPARGC1A binds to peroxisome proliferation-activated receptor γ (PPARγ) by induction of PPARγ ligands, coactivating PPARγ target genes, involved in antioxidant activity. Compared with controls, expression levels of PPARγ, PGC-1α and γ glutamylcysteine synthetase (γ-GCS) have been reported to be significantly increased in the lungs of patients with mild COPD, and progressively decreased in more severe disease.20 These authors concluded that γ-GCS showed compensatory upregulation in the early stage of COPD, which progressively decompensated with disease progression and that the activation of the PPARγ/PGC-1α pathway may protect against COPD progression by upregulating γ-GCS and relieving oxidative stress. Furthermore PPARGC1A has been described to be involved in bronchial smooth muscle remodelling and skeletal muscle wasting.21 ,22 For GSTO2, a strong association of the Asn142Asp SNP with FEV1 and FVC was found in the Framingham Heart Study.23 In a latter study, the Asn142Asp polymorphism in GSTO2 and the GSTO1 140Asp/GSTO2 142Asp haplotype were associated with increased risk of COPD but failed to reveal an association between lung function parameters and non-synonymous coding SNPs in the GSTO genes.24 Polymorphisms in GSTO2 were also associated with COPD either with or without lung cancer.25
Pathways involved in apoptosis and in handling of xenobiotic pathways were significantly enriched for causal genes. The results of the present study aid in understanding the underlying genetic dysregulation of apoptosis. In general, increased apoptosis of endothelial cells and fibroblasts has been shown to contribute to the development of emphysema.26 ,27 This is partly due to an imbalance caused by excess oxidant and protease effects related to cigarette smoking and to intrinsic dysregulation of apoptosis induced in susceptible individuals. This may explain why the apoptotic effects are larger in smokers with COPD than smokers without COPD. Dysregulation of apoptosis can also work the other way: decreased apoptosis in cells of the immune system can lead to sustained inflammation, and when occurring in fibroblasts (eg, in the bronchial wall) can induce fibrosis. Emphysema is characterised by alveolar cell apoptosis, which was shown to be mediated by increased levels of apoptotic proteins including TRAIL receptors.28 Interestingly, TRAIL signalling was the most significant pathway enriched with causality genes using Partek. TRAIL signalling has been shown to regulate immune responses in the lung and can lead to a sustained, proinflammatory response that contributes to vascular disease. An opposite effect is related to the death receptor signalling pathway, which, in humans, binds with TRAIL, induces formation of a death-inducing signalling complex, ultimately leading to caspase activation and initiation of apoptosis.29
The aryl hydrocarbon receptor signalling pathway is involved in xenobiotic clearance and therefore of relevance to the known vulnerability of patients with COPD to (cigarette) smoke. Furthermore additional metabolic pathways including the glutathione metabolism pathway and two subpathways involving metabolism of the cyanoamino acids, taurine and hypotaurine were also significantly enriched in the causal analysis. Genetic variants in a number of genes in the glutathione pathway have previously been associated with risk for COPD.25 ,30
No other large-scale lung eQTL dataset is available in patients with and without COPD. To replicate the most biologically relevant causality models, we relied on two genome-wide expression datasets. Causality genes were first validated in a gene expression study of bronchial airway epithelial cells obtained by bronchoscopy and then in a transcriptomic study of whole lung explanted at surgery. Whole genome genotyping is not available for these datasets. Accordingly, replication of significant triplets (ie, SNP–mRNA–phenotype) can only evaluate the concordance between gene expression and phenotype. Additional studies with phenotype, genotype and gene expression in the lung would be required to provide full validation.
In conclusion, integration of lung-specific eQTL data with GWAS from the same individuals has revealed interesting and potentially causal pathways of airflow obstruction. GWAS have revolutionised our ability to identify gene variants that contribute to susceptibility for common complex genetic diseases but often do not pinpoint the exact genes or mechanisms. Causal pathway analysis involving the joint examination of genetic and genomic data is a vital next step in discovering novel biomarkers and therapeutic targets in airflow obstruction as evidenced in the present study.
Acknowledgments
The authors would like to thank Christine Racine and Sabrina Biardel at the IUCPQ site of the Respiratory Health Network Biobank of the FRQS for their valuable assistance. They also acknowledge the staff at Calcul Québec for IT support with the high-performance computer clusters. At UBC the authors thank the biobank coordinator Dr Mark Elliott. At the Groningen UMCG site Marnix Jonker is thanked for his support in selecting, handling and sending of lung tissues. The authors thank Dr Joshua Millstein of University of Southern California for helpful advice on causality inference methodology.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table 1
Footnotes
ML, WT, and KH contributed equally.
-
Contributors WT, KH, YB, MLav, PDP, DDS and DSP were involved in the conception and design of the study. MLam, WT, KH, YB, MLav, KSt, JDC, CC, MC, KSh, JCH, C-AB, SL, MEL, ASp, PDP, DN and DSP were involved in the acquisition and/or the analysis and interpretation of the data. MLam, WT, KH, YB, MB, ASa, PDP, DDS and DSP contributed to the writing or the revision of the manuscript.
-
Funding This study was funded by Merck Research Laboratories, the Chaire de pneumologie de la Fondation JD Bégin de l'Université Laval, the Fondation de l'Institut universitaire de cardiologie et de pneumologie de Québec, the Respiratory Health Network of the FRQS, the Cancer Research Society and Read for the Cure, and the Canadian Institutes of Health Research (MOP-123369). YB was a research scholar from the Heart and Stroke Foundation of Canada and he is now recipient of a Junior 2 Research Scholar award from the Fonds de recherche Québec—Santé (FRQS). DDS is a Tier 1 Canada Research Chair for COPD. ML is the recipient of a doctoral studentship from the Fonds de recherche Québec - Santé (FRQS).
-
Competing interests CAB received a grant from Rosetta Merck. DN is a full-time employee of Merck. DSP received consultancy fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Takeda, TEVA and a grant from Chiesi. DDS has served on advisory boards of Almirall, Nycomed, Talecris, AstraZeneca, Merck Frosst, Novartis and GlaxoSmithKline, received grants from AstraZeneca, GlaxoSmithKline and Wyeth, and received honoraria for speaking engagements from Takeda, AstraZeneca, GlaxoSmithKline and Boehringer Ingelheim. JDC received consultancy fees from Metera Biosciences and Immuneering. MB received grants from TEVA, AstraZaneca and Chiesi. WT received a grant from Merck Sharp Dohme, received consultancy fees from Pfizer, and received lecture fees from GlaxoSmithKline, Chiesi and Roche Diagnosis.
-
Patient consent Obtained.
-
Ethics approval Institutional Review Board guidelines at the three sites.
-
Provenance and peer review Not commissioned; externally peer reviewed.