Article Text

Abstract
Background We hypothesised that increased oxidative stress, as present in the airways of asthma and chronic obstructive pulmonary disease (COPD) patients, induces epithelial damage and reduces epithelial responsiveness to suppressive effects of corticosteroids on proinflammatory cytokine production and barrier function.
Methods We induced oxidative stress by H2O2 and/or cigarette smoke extract (CSE) in human bronchial epithelial 16HBE cells and primary bronchial epithelial cells (PBEC) derived by brushings from asthma patients, COPD patients, and smoking and non-smoking control individuals. We investigated effects of budesonide on barrier function (electrical resistance) and TNF-α-induced proinflammatory cytokine production (IL-8/CXCL8, granulocyte macrophage-colony stimulating factor (GM-CSF)).
Results We observed that H2O2 and CSE reduce epithelial resistance. Budesonide significantly counteracted this effect, likely by protection against epidermal growth factor receptor-dependent cell-cell contact disruption. Furthermore, budesonide suppressed proinflammatory cytokine production. H2O2 pretreatment reduced this effect of budesonide on cytokine production in both 16HBE cells and PBECs. Importantly, PBECs from asthma and COPD patients were less sensitive to budesonide with respect to cytokine production and barrier function than PBECs from control subjects.
Conclusions Together, our data indicate that budesonide suppresses epithelial proinflammatory responses and barrier dysfunction and that oxidative stress reduces these effects in airway epithelium from asthma and COPD patients. Therefore, restoration of corticosteroid responsiveness in asthma and COPD may act to improve the airway epithelial barrier.
- Airway Epithelium
- Asthma
- COPD Pharmacology
- GM-CSF
- Oxidative Stress
Statistics from Altmetric.com
Key messages
What is the key question?
-
The main question is whether oxidative stress can reduce corticosteroid responsiveness in airway epithelium and how this affects the regulation of epithelial barrier function and cytokine production.
What is the bottom line?
-
We show for the first time that budesonide protects against oxidative stress-induced epithelial barrier dysfunction, and that oxidative stress decreases epithelial responsiveness to budesonide.
Why read on?
-
This may have important implications for asthma and COPD, since we also show that epithelial cells from asthma and COPD patients are less responsive to budesonide.
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are both chronic inflammatory respiratory disorders with a worldwide increase in incidence. Inhaled corticosteroids (ICS) are the cornerstone of asthma treatment, with a broad anti-inflammatory spectrum. Nevertheless, approximately 20% of the asthma patients are insensitive to ICS and fail to respond even when higher ICS doses are given. Furthermore, ICS provide relatively little therapeutic benefit in COPD. They reduce exacerbations, but do not effectively change the course of the disease in the majority of COPD patients.
Cigarette smoking is the main risk factor for both the development and progression of COPD, and is, additionally, associated with airway remodelling, accelerated lung function decline and more severe disease in asthma.1 ,2 Airway epithelium forms the first, tightly regulated barrier against environmental insults, including cigarette smoke, and is a source of proinflammatory cytokines, especially when damaged.3 Aberrant epithelial damage and repair is thought to play a central role in the pathogenesis of both asthma and COPD.4–7 In asthma, disrupted airway epithelial integrity is frequently observed, with loss of junctional proteins that maintain barrier function, for example, E-cadherin and zona occludens (ZO)-1.3 ,6 ,8 Oxidative stress, as induced by inhaled cigarette smoke,9 inflicts epithelial damage and promotes the release of cytokines that attract and activate neutrophils, including IL-8/CXCL8 and granulocyte macrophage-colony stimulating factor (GM-CSF).3 Additionally, the generation of reactive oxygen species (ROS) by neutrophils and/or macrophages may lead to increased oxidative stress in asthma and COPD.10 ,11 This may have clinical relevance, since increased ROS production by neutrophils has been associated with more severe asthma and COPD.12
In addition to epithelial damage, oxidative stress has been implicated in corticosteroid unresponsiveness13 ,14 and may contribute to the poorer response to corticosteroids observed in smoking compared with non-smoking asthmatics.13 ,15 Corticosteroids have been reported to promote regeneration of airway and corneal epithelium.16 ,17 It is currently still unknown whether oxidative stress can induce corticosteroid unresponsiveness in airway epithelium and how this affects the regulation of epithelial barrier function and cytokine production. Therefore, we studied the effect of oxidative stress on epithelial barrier function, cytokine production and responsiveness to the clinically relevant corticosteroid budesonide. Furthermore, we studied whether the protective effects of budesonide are reduced in epithelial cells from asthma and COPD patients.
Methods
Culture of human bronchial epithelial cells
The human bronchial epithelial cell line 16HBE was kindly provided by Dr DC Gruenert (University of California, San Francisco, California, USA) and cultured in EMEM medium/10% FCS (Biowhittaker, Verviers, Belgium) as described previously.18 Primary bronchial epithelial cells (PBEC) were obtained from bronchial brushings in five asthma patients, six current smokers with COPD GOLD stage II, six ex-smokers with COPD GOLD stage IV, eight current smoking control individuals and 14 non-smoking control individuals (see table 1). With the exception of the COPD stage IV patients, subjects did not use ICS, long-acting β-agonists and long-acting anticholinergics for at least 4 weeks preceding the study. The Medical Ethics Committee of the University Hospital of Groningen approved the study and all participants gave their written informed consent. Cells were cultured, as described previously,19 in bronchial epithelium growth medium (BEGM, Lonza) on collagen/fibronectin-coated flasks, stored in liquid nitrogen and used for experiments at passage 3.
Characteristics of the subjects
Stimulation of the cells
Cells were grown in duplicates in 24-well plates or on electrical cell-substrate impedance sensing (ECIS) arrays for 3–4 days until 80–90% confluency and subsequently serum-deprived (16HBE) or cultured in bronchial epithelial cell basal medium (BEBM, Lonza) with transferrin and insulin (PBECs) overnight. Cells were pretreated with H2O2 (100 µM) for 2 h, followed by budesonide (1, 10 and 100 nM) for 2 h and subsequently stimulated with TNF-α (10 ng/ml)20 for 24 h. Supernatants were collected or cells were harvested for RNA isolation. For ECIS experiments, cells were pretreated with budesonide (1–100 nM) for 2 h and subsequently exposed to vehicle (medium), H2O2 (100 µM), cigarette smoke extract (CSE) (5–7.5%) or epidermal growth factor (EGF) (10 ng/ml).2,1 See online data supplement for the methods of immunodetection and ECIS.
Preparation of CSE
CSE was prepared as described previously.22 In short, Kentucky 3R4F research-reference cigarettes (The Tobacco Research Institute, Lexington, Kentucky, USA) were used without filter. Smoke from two cigarettes was bubbled through 25 mL medium (100% CSE). The extract was prepared within 15 min prior to use.
Measurement of IL-8 and GM-CSF secretion levels
Protein levels were measured in cell-free supernatants using ELISA kits according to the manufacturer's guidelines (R&D Systems Europe, Abingdon, UK).
Statistics
Data were analysed using Student t test for paired differences within the cell line, two-way analysis of variance (ANOVA) for time curves in the cell line (ECIS), Wilcoxon-signed rank test for paired observations within subject groups, Mann–Whitney test for analyses between subject groups, and one-way ANOVA for budesonide concentration series within subject groups.
Results
Oxidative stress reduces the inhibitory effect of corticosteroids on GM-CSF, but not IL-8 production in human bronchial epithelium
We first examined the effect of pretreatment of healthy human bronchial epithelium with the oxidant H2O2 (2 h) on proinflammatory cytokine release. We stimulated the cells with TNF-α (24 h) as a relevant mediator of respiratory inflammation in asthma and COPD.23–25 Both TNF-α and H2O2 significantly induced IL-8 secretion in 16HBE cells, which was dose-dependently inhibited by budesonide to a similar extent. Pretreatment with H2O2 significantly increased the TNF-α-induced IL-8 secretion and significantly reduced the inhibitory effect of budesonide on TNF-α-induced IL-8 secretion (data not shown). In PBECs from healthy non-smoking donors, TNF-α substantially increased IL-8 (figure 1A,B) and GM-CSF (figure 1C) secretion. Budesonide dose-dependently inhibited baseline and TNF-α-induced IL-8 secretion, with the strongest effect of 100 nM budesonide, although this effect was not significantly different from 10 nM budesonide (figure 1A). By contrast with 16HBE cells, pretreatment with H2O2 did not further increase IL-8 or GM-CSF secretion (figure 1B,C), and the inhibitory effect on IL-8 remained present upon pretreatment with H2O2 (figure 1B). On the contrary, H2O2 pretreatment significantly reduced the inhibitory effect of budesonide on GM-CSF in PBECs (figure 1C).

Pretreatment with H2O2 reduces sensitivity of GM-CSF production to budesonide (BUD) in primary bronchial epithelial cells (PBEC) from healthy non-smokers (n=6–7). IL-8 and GM-CSF were measured in cell-free supernatants of (A) PBECs treated with 1–100 nM BUD for 2 h and subsequently incubated with/without TNF-α for 24 h and (B, C) PBECs pretreated with/without H2O2 for 2 h, followed by 100 nM BUD and subsequent stimulation with TNF-α for 24 h. Absolute IL-8 levels (pg/mL) and levels related to the values in absence of BUD are presented (A,B). Absolute GM-CSF levels (pg/mL) and levels related to the values in the absence of BUD (C). *=p<0.05 for the values with BUD compared with its absence or between the indicated values.
Reduced responsiveness to budesonide in PBECs from asthma and COPD patients compared with control PBECs
Next, we determined whether an increased oxidant burden in the airways may result in reduced corticosteroid responsiveness of cultured bronchial epithelial cells. We compared ICS responsiveness of PBECs from asthma patients to non-smoking controls and PBECs from GOLD stage II COPD patients to age-matched smokers and non-smokers with normal lung function. Exposure to TNF-α resulted in a significant increase in IL-8 and GM-CSF release in all subject groups (figure 2A), with no significant differences in TFN-α-induced IL-8 or GM-CSF levels between the subject groups. Budesonide inhibited proinflammatory cytokine production in all groups, inducing a significant reduction in IL-8 and GM-CSF production when used in a concentration of 100 nM, with the strongest effect of 41(26–71)% GM-CSF inhibition and 41(31–59)% IL-8 inhibition in PBECs from non-smoking controls (figure 2B). Of note, the inhibitory effect of budesonide on GM-CSF was significantly less in PBECs from asthma patients than from healthy non-smoking controls (figure 2B). Similar results were observed for IL-8, although the inhibitory effect of 10 nM budesonide only tended to be decreased in PBECs from asthma patients. The inhibitory effect of 100 nM budesonide on GM-CSF was significantly less in PBECs from COPD patients than from non-smoking controls (figure 2B). On the contrary, no significant differences in the inhibitory effect of budesonide on IL-8 were observed between PBECs from COPD patients, smoking and non-smoking controls (figure 2B). Furthermore, the effects of budesonide on both IL-8 and GM-CSF production were not significantly different between the smoking and non-smoking groups (figure 2B). Together, these data indicate that PBECs from asthma and COPD patients, but not those from control smokers, are less responsive to budesonide than PBECs from healthy non-smokers, in particular regarding GM-CSF production.

Sensitivity of IL-8 and GM-CSF production to budesonide (BUD) in primary bronchial epithelial cells (PBECs) from asthma patients (n=5), healthy controls (n=13), COPD patients (n=6), smoking controls (n =8) and non-smoking controls (n=7). PBECs were pretreated with/without BUD for 2 h (1–100 nM) and subsequently stimulated with TNF-α. IL-8 and GM-CSF were measured in cell-free supernatants. (A) Absolute baseline and TNF-α-induced values of IL-8 and GM-CSF (pg/mL). (B) IL-8 and GM-CSF levels related to the values in absence of BUD. *=p<0.05, **=p<0.01 and ***=p<0.001 for the values with BUD compared with its absence, or between the indicated values.
Budesonide improves the restoration of epithelial cell-cell contacts upon exposure to oxidative stress
We also studied whether oxidative stress and changes in responsiveness to budesonide may have consequences for epithelial barrier function. Addition of H2O2 (100 µM) caused transient epithelial barrier dysfunction in a monolayer of 16HBE cells, as reflected by a decrease in low-frequency resistance (figure 3A). H2O2 treatment did not affect the high-frequency capacitance (data not shown), the most sensitive parameter for changes in cell-matrix attachment,26 indicating that H2O2 mainly affects intercellular contacts. When used in a higher concentration (1 mM), H2O2 induced a dramatic decrease in low-frequency resistance and increase in high-frequency capacitance back to the levels of the uncovered electrode, reflecting detachment of the cells, from which the cells were not able to recover (data not shown). Therefore, we decided to use 100 µM H2O2 in our further experiments. Treatment with budesonide alone (10 nM) did not significantly increase resistance (figure 3A). Of interest, budesonide improved H2O2-induced epithelial barrier dysfunction, with a significant increase in resistance at 6, 12 and 24 h (figure 3A). Next, we examined the effect of 7.5% CSE, which induced robust epithelial barrier dysfunction (figure 3B), without causing cell detachment from the matrix and/or cell death.4 Importantly, pretreatment with 10 nM budesonide fully counteracted the CSE-induced decrease in barrier function (figure 3B).

Budesonide (BUD) protects against oxidative stress-induced epithelial barrier dysfunction and promotes the restoration of cell-cell contacts upon wounding. 16HBE cells were pretreated for 2 h with/without BUD) at 96 h and subsequently exposed to H2O2, cigarette smoke extract (CSE, 7.5%), epidermal growth factor (EGF) or wounding by electroporation. Resistance was measured at 400 Hz. (A) BUD (10 nM) was added 2 h prior to H2O2. Values were normalised to the levels at 3 h after the addition of H2O2 (representative experiment) and the area-under-the-curve (AUC, Mean±SEM, n=4) was calculated over 12 h, the time period in which H2O2 decreased in barrier function. (B) BUD (10 nM) was added 2 h prior to CSE. Values were normalised to the levels at 2 h prior to CSE addition (representative experiment) and the AUC (Mean±SEM, n=4) calculated over 24 h, starting from the time-point 2 h prior to CSE addition. (C) Absolute values (representative experiment) and values normalised to the levels at 2 h prior wounding (Mean±SEM, n=5). (D) BUD (100 nM) was added 2 h prior to EGF. Values were normalised to the levels just prior to EGF addition (representative experiment) and AUC values calculated over 24 h starting from the time-point 2 h prior EGF addition (Mean±SEM, n=5). *=p<0.05 and **=p<0.001 between the indicated values.
Budesonide improves the restoration of epithelial cell-cell contacts upon wounding by electroporation and exposure to EGF
Additionally, we studied the effect of budesonide on epithelial regeneration upon wounding by electroporation, resulting in complete cell detachment, followed by a first phase of barrier recovery due to cell migration and a second phase due to cell-cell contact restoration.26 Budesonide did not affect the cell migration response during the first phase, but dose-dependently increased epithelial resistance during the second phase of the wound-healing response (figure 3C). Together, our findings indicate that budesonide specifically improves the formation of cell-cell contacts upon epithelial damage.
We previously reported that EGFreceptor (EGFR) activation reduces cell-cell contact restoration upon wounding and is involved in CSE-induced barrier dysfunction in 16HBE cells as well as PBECs, inducing degradation of ZO-1.4 Furthermore, we observed that EGFR is involved in house dust mite (HDM)-induced barrier dysfunction21 and that HDM induces EGFR phosphorylation in PBECs from asthma patients.27 Therefore, we studied the effect of EGF and observed that the addition of EGF induces a similar decrease in epithelial barrier function as CSE, and this was also counteracted by budesonide (100 nM, figure 3D). Moreover, budesonide was able to inhibit EGFR phosphorylation at Tyr1174 within 30–240 min (data not shown), with an optimal effect at 240 min (figure 4A). In line with these and our previous data, budesonide partially prevented the CSE-induced reduction in ZO-1 levels, whereas it did not increase baseline levels of ZO-1 (figure 4B). Together, these results indicate that budesonide exerts protective effects on cell-cell contacts in 16HBE cells, which likely involves the inhibition of EGFR activity.
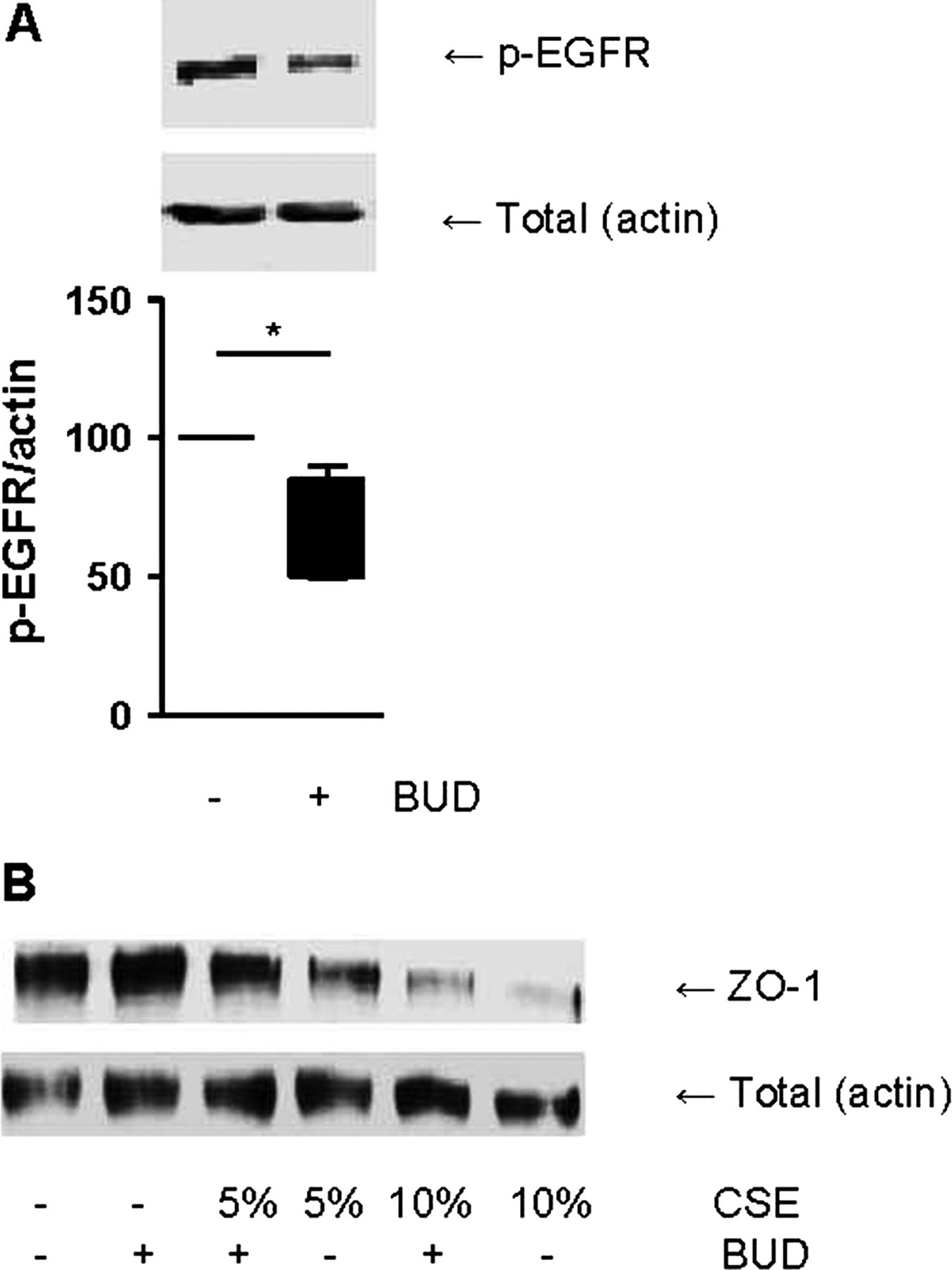
Budesonide (BUD) reduces the phosphorylation of epidermal growth factor receptor (EGFR) and attenuates the cigarette smoke extract (CSE)-induced reduction in zona occludens (ZO)-1 levels. (A) 16HBE cells were treated with/without BUD (100 nM) for 4 h. Total cell lysates were prepared. Phospho-EGFR was detected by western blotting and actin was used as loading control. A representative of 5 independent experiments is shown. Densitometry was performed, and the corresponding graph shows the phospho-EGFR levels related to actin (Mean±SEM, n=5). (B) 16HBE cells were treated with/without BUD (100 nM) for 2 h followed by 24 h exposure to CSE (5% or 10%). ZO-1 and E-cadherin were detected by western blotting and actin was used as loading control. A representative of three independent experiments is shown. *=p<0.05 between the indicated values.
Budesonide improves epithelial barrier function in control PBECs, and this effect is reduced in PBECs from asthma and COPD patients
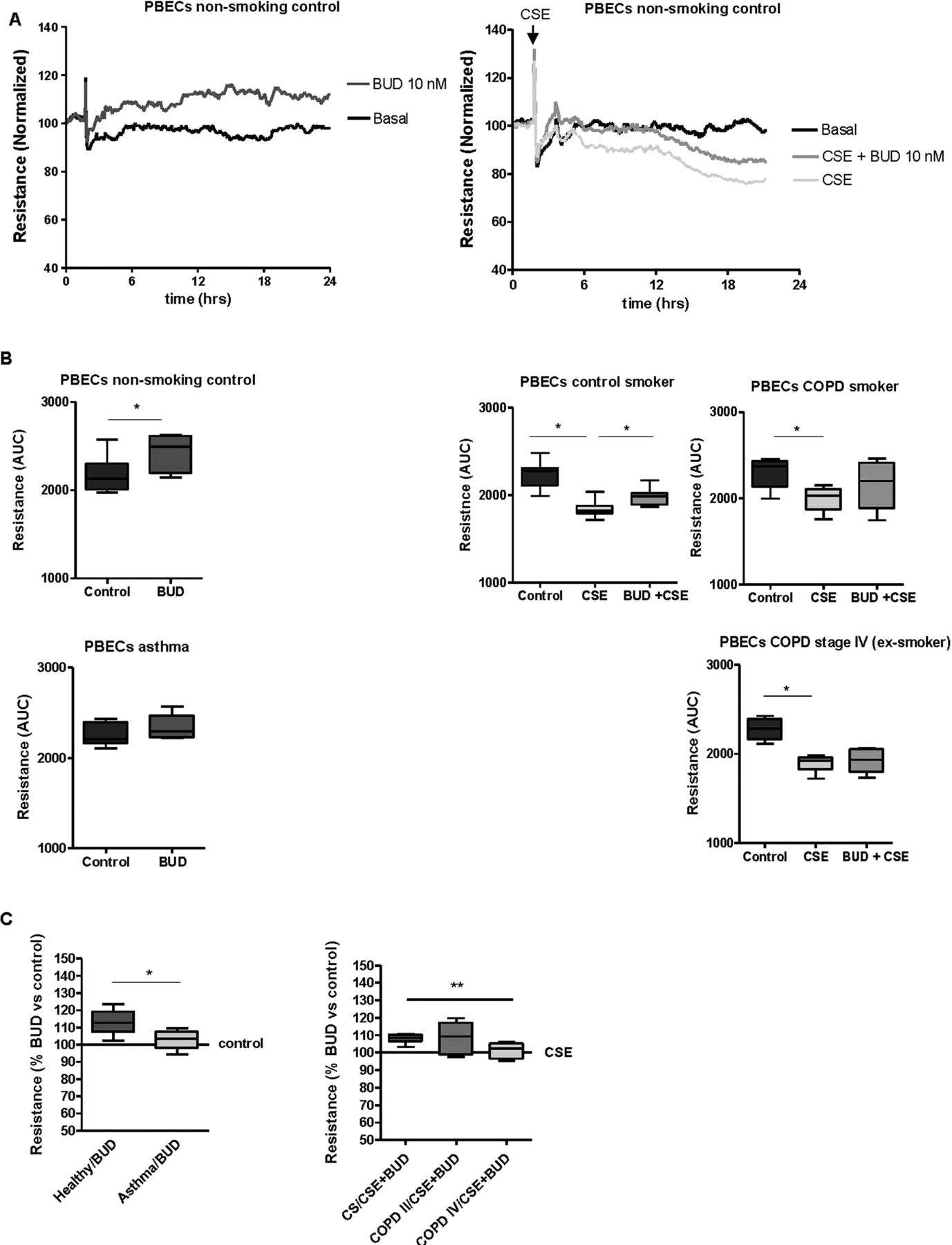
We further assessed whether budesonide also improves barrier function in PBECs from asthma patients, healthy non-smoking controls, COPD patients and age-matched control smokers and non-smokers. As previously observed,26 PBEC monolayers displayed approximately fourfold lower resistance levels than 16HBE monolayers. A similar effect of budesonide, as in 16HBE cells, was observed upon wounding of the PBEC monolayers (see online supplementary figure S1), although this failed to reach statistical significance. By contrast with 16HBE cells, budesonide (10 nM) significantly increased baseline resistance in PBECs from healthy non-smoking individuals (figure 5A). This effect was not present in asthma epithelium, with a significantly lower increase in resistance in the presence of budesonide compared with PBECs from healthy individuals (figure 5B). This indicates that the protective effect of budesonide on epithelial barrier function is compromised in asthma patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of budesonide (BUD) on epithelial barrier function in primary bronchial epithelial cells (PBECs) from non-smoking controls (n=7), asthma patients (n=5), control smokers (n=7), COPD stage II patients (n=6) and COPD stage IV patients (n=6). PBECs were pretreated for 2 h with/without BUD (10 nM) and subsequently incubated with/without 5% cigarette smoke extract (CSE). Resistance was measured at 400 Hz for 24 h and levels were normalised to the values just prior to the addition of BUD. (A) Absolute resistance values in a representative experiment in PBECs from a non-smoking individual. (B) The area-under-the-curve (AUC) was calculated over 24 h starting from the time-point just prior to addition of BUD and the median (range) values per subject group are shown. (C) The upregulatory effect of BUD is presented as percentage of the AUC values in the control or CSE-treated PBECs (as indicated) for the different subject groups; *=p<0.05 and **=p<0.001 between the indicated values.
In accordance with our previous data,4 CSE induced a similar and significant decrease in resistance in PBECs from non-smokers (figure 5A), control smokers and smokers with COPD GOLD stage II (figure 5C), with a maximal effect around 24 h. While pretreatment with 100 nM budesonide had no significant effect (data not shown), pretreatment with 10 nM budesonide partially protected against CSE-induced barrier dysfunction in PBECs from smoking and non-smoking controls (figure 5A,B). By contrast, budesonide did not significantly improve CSE-induced barrier dysfunction in PBECs from COPD GOLD stage II patients. Furthermore, the protective effect of budesonide was completely absent in PBECs from COPD GOLD stage IV patients, its effect being significantly lower than in control smokers (figure 5C). These data indicate that epithelial cells from asthma and COPD patients, particularly those with very severe COPD, are less sensitive to ICS with respect to their barrier function.
Discussion
We demonstrate that oxidative stress induces epithelial barrier dysfunction and corticosteroid unresponsiveness of pro-inflammatory cytokine production in bronchial epithelial cells. Since cigarette smoking and respiratory inflammation may increase the oxidant burden in asthma and COPD, our results may have important clinical relevance. Our data show that GM-CSF secretion is less sensitive to budesonide in PBECs from asthma and COPD patients than from healthy non-smoking individuals. Furthermore, we demonstrate for the first time that budesonide counteracts oxidative stress-induced barrier dysfunction in human bronchial epithelium, and that this protective effect is also impaired in PBECs from asthma and COPD patients, particularly in patients with very severe COPD. We did not observe this in PBECs of control smokers, indicating a disease-related defect in corticosteroid responsiveness.
A possible explanation may be that healthy airway epithelium displays a higher capacity to compensate for oxidative stress by more adequate antioxidant responses, rendering it less prone to reduced corticosteroid sensitivity. Accordingly, an increased oxidant burden due to lower levels of antioxidant defence has been reported in COPD, and this was associated with chronic airflow limitation.28 Additionally, we observed reduced corticosteroid responsiveness in epithelium from asthma patients. This cannot be an effect that involves cigarette smoking, since none of the asthma patients were smokers. Nevertheless, it has been well accepted that oxidative stress contributes to the pathogenesis of asthma. Inflammatory cells in the airways of asthma patients release oxidants, and inflammatory cells recruited to the airways have been described to possess an exceptional capacity to produce oxidants in both asthma and COPD.29 Since we show that H2O2 reduces epithelial responsiveness to budesonide, we anticipate that the observed reduction in corticosteroid responsiveness of airway epithelial cells from both asthma and COPD patients may result from increased oxidant levels in the airways. The further reduction in corticosteroid responsiveness in epithelium from very severe COPD patients may derive from more severe airway inflammation, since pack-years of smoking were similar between COPD GOLD stage II and IV patients. On the other hand, we can currently not exclude that ICS use contributes to the epithelial corticosteroid unresponsiveness in the GOLD stage IV patients, the only group in our study allowed to use ICS.
Oxidative stress may reduce corticosteroid sensitivity by mechanisms involving phosphorylation of the glucocorticoid receptor (GR) as well as phosphorylation and inactivation of histone deacetylatases.30 ,31 GR function is regulated by acetylation, and its deacetylation is necessary for gene repression, for example, through binding to the proinflammatory transcriptional factors NF-κB. Furthermore, histone deacetylation is a well established negative regulator of NF-κB and the transcription factor AP-1, and corticosteroids recruit deacetylases to the promoter of proinflammatory genes.30 Acetylated NF-κB has been reported to preferentially bind to the IL-8, but not GM-CSF gene promoter,32 suggesting that changes herein would rather affect IL-8 than GM-CSF responsiveness, which is not in line with our results. Instead, the transcriptional regulation of GM-CSF by corticosteroids may involve GR binding to the NF-AT/AP-1 sites in the distal enhancer.33 The mechanisms involved in the reduced responsiveness of GM-CSF production to corticosteroids in airway epithelial cells will be studied in the future.
In line with our findings, it has been reported that GM-CSF release from peripheral blood mononuclear cells of asthma patients is less sensitive to inhibition by corticosteroids than that of healthy controls.34 GM-CSF promotes the survival, proliferation and differentiation of granulocytes. The relative corticosteroid unresponsiveness of neutrophils has been proposed to contribute to the inefficient control of airway inflammation in corticosteroid refractory asthma. However, this does not explain the corticosteroid insensitivity of neutrophilic airway inflammation in asthma, since inhibition of GM-CSF release from resident cells should still be able to suppress neutrophil survival. Thus, the oxidant-induced unresponsiveness of airway epithelial GM-CSF release may constitute a novel mechanism underlying corticosteroid insensitive neutrophilic inflammation in asthma as well as COPD.
In addition to the effects on GM-CSF, our data suggest that reduced corticosteroid responsiveness may have important consequences for epithelial integrity. We show that budesonide is able to improve barrier function of healthy epithelium. This capacity was reduced in PBECs from asthma and COPD patients at baseline and in the presence of CSE, respectively. With respect to the mechanism involved in the protective effect of corticosteroids on epithelial barrier function, we have previously shown that CSE induces activation of the EGFR and subsequent disruption of epithelial junctions. We report here that budesonide also counteracts EGF-induced barrier dysfunction and inhibits EGFR phosphorylation in 16HBE cells. Of interest, and requiring further investigation, a distinct membrane receptor has been implicated in the rapid effects of corticosteroids on membrane kinases, for example, extracellular signal-regulated kinase, that may also regulate EGFR phosphorylation.
In line with our findings, Sekiyama et al35 have shown that the corticosteroid dexamethasone increases transepithelial resistance of epithelial Calu-3 cells during 7-day culture. By contrast with a role for the inhibition of EGFR activity, the authors imply a role for EGFR activation by corticosteroids in this effect. They show that EGFR siRNA impairs epithelial barrier function and that dexamethasone increases the level of EGFR phosphorylation. This effect predominantly occurred at the Tyr922 residue, however, Sekiyama et al did not study phosphorylation at the major auto-activation residue Tyr1174, which we found to be reduced upon budesonide treatment. The discrepancy between our studies may further derive from the use of the Calu-3 cell line that originates from submucosal glands, while we used bronchial epithelial cells. Finally, Sekiyama and coworkers analysed transepithelial resistance by conventional measurements using a voltohmmeter, which is not able to distinguish between cell-cell and cell-matrix contacts.26
In conclusion, we show that an increased oxidant burden, as observed in asthma and COPD, may lead to reduced corticosteroid responsiveness in airway epithelium, with important consequences for the integrity of the epithelial barrier and associated production of pro-inflammatory cytokines. Strategies to reduce oxidative stress and increase corticosteroid responsiveness may lead to improved epithelial barrier function as well as reduced proinflammatory epithelial responses in asthma and COPD.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
*DP and MvdB contributed equally.
-
Contributors All authors critically reviewed the manuscript and finally approved it. IH was involved in conception and design of the study, analysis and interpretation of data and drafting the article. NK, MJ and RH were involved in data collection, analysis and interpretation. ET, KK and D-JS were involved recruitment and inclusion of patients. AvO, NtH, DP and MvdB were involved in design and conception of the study and revision of the manuscript.
-
Funding This research was supported by AstraZeneca (grant number: SEML-89KDE8) and partially supported by and performed within the framework of the Top Institute Pharma project T1–201 “COPD, transition of systemic inflammation into multi-organ pathology”, with partners University Medical Center Groningen, University Medical Center Utrecht, University Medical Center Maastricht, Nycomed BV, GlaxoSmithKline, Danone, AstraZeneca, and Foundation TI Pharma.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Medical Ethics Committee of the University Hospital of Groningen.
-
Provenance and peer review Not commissioned; externally peer reviewed.