Article Text

Abstract
Rationale Arikace is a liposomal amikacin preparation for aerosol delivery with potent Pseudomonas aeruginosa killing and prolonged lung deposition.
Objectives To examine the safety and efficacy of 28 days of once-daily Arikace in cystic fibrosis (CF) patients chronically infected with P aeruginosa.
Methods 105 subjects were evaluated in double-blind, placebo-controlled studies. Subjects were randomised to once-daily Arikace (70, 140, 280 and 560 mg; n=7, 5, 21 and 36 subjects) or placebo (n=36) for 28 days. Primary outcomes included safety and tolerability. Secondary outcomes included lung function (forced expiratory volume at one second (FEV1)), P aeruginosa density in sputum, and the Cystic Fibrosis Quality of Life Questionnaire—Revised (CFQ-R).
Results The adverse event profile was similar among Arikace and placebo subjects. The relative change in FEV1 was higher in the 560 mg dose group at day 28 (p=0.033) and at day 56 (28 days post-treatment, 0.093L±0.203 vs −0.032L±0.119; p=0.003) versus placebo. Sputum P aeruginosa density decreased >1 log in the 560 mg group versus placebo (days 14, 28 and 35; p=0.021). The Respiratory Domain of the CFQ-R increased by the Minimal Clinically Important Difference (MCID) in 67% of Arikace subjects (560 mg) versus 36% of placebo (p=0.006), and correlated with FEV1 improvements at days 14, 28 and 42 (p<0.05). An open-label extension (560 mg Arikace) for 28 days followed by 56 days off over six cycles confirmed durable improvements in lung function and sputum P aeruginosa density (n=49).
Conclusions Once-daily Arikace demonstrated acute tolerability, safety, biologic activity and efficacy in patients with CF with P aeruginosa infection.
- Respiratory Infection
- Cystic Fibrosis
- Bacterial Infection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
To examine the safety and efficacy of 28 days of once-daily Arikace in cystic fibrosis (CF) patients chronically infected with Pseudomonas aeruginosa.
What is the bottom line?
-
Our data provide evidence for the short-term safety, tolerability, biologic activity and efficacy of nebulised Arikace in CF patients chronically infected with P aeruginosa.
Why read on?
-
Our results with Arikace may contribute to novel utilisation of topical antibiotics to treat Pseudomonas infection in CF patients.
Introduction
Cystic fibrosis (CF) is a life-shortening genetic disease affecting over 70 000 CF patients worldwide. CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene.1 ,2 Morbidity and mortality associated with chronic Pseudomonas aeruginosa lung infection affects approximately 80% of CF patients by adulthood.3 ,4 Although chronic P aeruginosa infection accelerates the loss of lung function and is an independent contributor to mortality,5 more recent studies have reported reduced effects of Pseudomonas infection on long-term patient outcomes.6 ,7
Inhaled antibiotics, including aztreonam for inhalation solution (AI), colistimethate sodium dry powder (Colobreathe), and tobramycin inhalation solution (TIS) are approved for use in CF patients with chronic P aeruginosa infection.8–13 All three reduce bacterial density in CF sputum, stabilise lung function, and have become important components of CF care. TIS and AI are Federal Drug Administration (FDA) approved for use in the USA, while TIS, AI and Colobreathe dry powder inhaler are approved in Europe. Recent studies examining cycled use of AI over 18mos in CF patients chronically infected with P aeruginosa indicate that lung function improvements gained during 28 days of treatment are typically 3–5% above baseline values after the first three treatment cycles, with decline towards baseline off-treatment.11 ,14 Similar findings have been reported with TIS, with <5% improvements in lung function relative to baseline observed after the first treatment cycle.15 Two recent head-to-head open-label trials of AI compared with TIS, and colistimethate sodium dry powder compared with TIS have shown that their effects on FEV1 are small, particularly following three treatment cycles.13 ,16 This may reflect increased use of anti-Pseudomonas antibiotics in the current era of CF care, highlighting the need for novel approaches to treat Pseudomonas infection.
Arikace is a unique formulation that encapsulates aqueous amikacin in neutral liposomes composed of dipalmitoyl-phosphatidylcholine (DPPC) and cholesterol, and is delivered to patients using an optimised investigational eFlow Nebuliser System.17 Upon nebulisation, Arikace liposomes (∼300 nm) penetrate CF sputum, are lysed by local P aeruginosa-derived products, and have a prolonged lung half-life (several hours) relative to liposome-free antibiotics.18 ,19 Arikace reduces P aeruginosa density in animal models of lung infection, and studies in preclinical model systems and in human subjects support once-daily dosing.20 ,21
In this report, we describe the safety, tolerability, efficacy and pharmacokinetics of four doses of once-daily Arikace for 28 days compared with placebo in CF patients chronically infected with P aeruginosa.
Methods
The Phase II Arikace programme included randomised, double-blind, placebo-controlled, multiple-dose, multicentre trials in subjects with CF. Parallel studies were conducted in Europe (13 sites) and the USA (19 sites). Institutional Review Board, IEC approvals and informed consents were obtained at each site, and for all study subjects. The studies were performed in accordance with International Conference on Harmonisation, Good Clinical Practice guidelines, and the Declaration of Helsinki. Pooled analyses of common endpoints between the placebo-controlled European and US studies were established a priori (safety, adverse events (AE), serious AE (SAE), lung function, CFQ-R, microbiology and pharmacokinetics).
Subjects
Inclusion criteria included a CF diagnosis (sweat Cl– >60 mmol/L or two CF-associated mutations with organ system manifestation of CF), age ≥6 yrs, FEV1 ≥40% predicted, chronic P aeruginosa infection (four positive cultures over 2 years, including one within 3 months before screening and one at screening), clinical stability off of inhaled, or intravenous antibiotics 28 days before dosing, and no known allergy to amikacin. Additional information regarding study subjects is found in the online supplement.
Study design
The preclinical programme for Arikace development provided safety and efficacy coverage for dosing up to 560 mg once daily (multiple 28-day cycles). In Europe, subjects were randomised to once-daily Arikace (280 mg or 560 mg) or placebo for 28 days, followed by 28 days off study drug. Cohort 1 subjects were randomised to 280 mg or placebo, and cohort 2 subjects to 560 mg or placebo, each in a 2 : 1 ratio (equal randomisation for Arikace study arms and combined placebo). Based on initial FDA recommendations for a dose-ascending study design, patients in the USA were equally randomised to once-daily Arikace (70 mg or 140 mg) or placebo. After enrolling 19 subjects, a prespecified Data Safety Monitoring Board (DSMB) safety review (coupled with examination of preliminary safety and efficacy data from the European trial) recommended amending the US protocol to dosing with 560 mg Arikace versus placebo (1 : 1 randomisation). Following subsequent FDA approval, the US study examined 560 mg Arikace nebulised once daily versus placebo, with postdosing monitoring for 56 days (weekly visits for 28 days of treatment, then visits at days 42 and 56 off study drug). To provide complete data on safety and tolerability, data from all dose groups in both trials are included. Open-label extension details are in the online supplement.
Endpoints
The primary study objective was safety and tolerability (including vital signs, predose and postdose lung function (acute tolerability), pulse oximetry, AEs, haematology, clinical chemistry, urinalysis, physical examinations, audiology and ECG and chest x-rays as needed). Secondary objectives are described in the online supplement.
Pharmacokinetic Analysis is included in the online supplement.
Statistical analyses of safety data
Baseline characteristics are described using summary statistics. Incidence of AEs by MedDRA system organ class and preferred term are displayed by treatment group. If an AE was reported more than once during the study period for a given patient, the greatest severity and the worst-case attribution are presented in the summary tables. Fisher's exact tests were used to compare AE rates between the dose cohorts and placebo cohort.
Differences in treatment arms for predose measurements of pulmonary function over time were compared using repeated measures ANOVA (rANOVA), adjusting for bronchodilator use. Treatment differences with respect to the changes from baseline to each measured study day in log10CFU/g, were similarly computed using ANOVA with no adjustments made for multiple comparisons. A non-parametric correlation of change in log10CFU/g with baseline minimal inhibitory concentration (MIC) was computed for each treatment group and displayed graphically by National Committee for Clinical Laboratory Standards-Susceptible, Intermediate, Resistant (NCCLS-SIR) categories of baseline MIC. Treatment differences in absolute and relative change in CFQ-R domain scores were computed using analysis of covariance (ANCOVA), adjusting for baseline scores; the proportion of patients with minimum clinically important difference on the respiratory scale by treatment group were compared by treatment group using a χ2 statistic. Statistical analyses were performed using SAS V.9.1 and S-PLUS 2000.
The study was adequately powered for the primary objective (safety) and secondary objectives. A minimum of 20 subjects were required in any cohort to have an 80% chance of detecting any AE if the underlying rate was 8% or higher. Analyses were performed on all subjects who received at least one dose of study drug (safety population).
Additional statistical analyses, including efficacy outcome measures, are included in the online supplement.
Results
Subjects
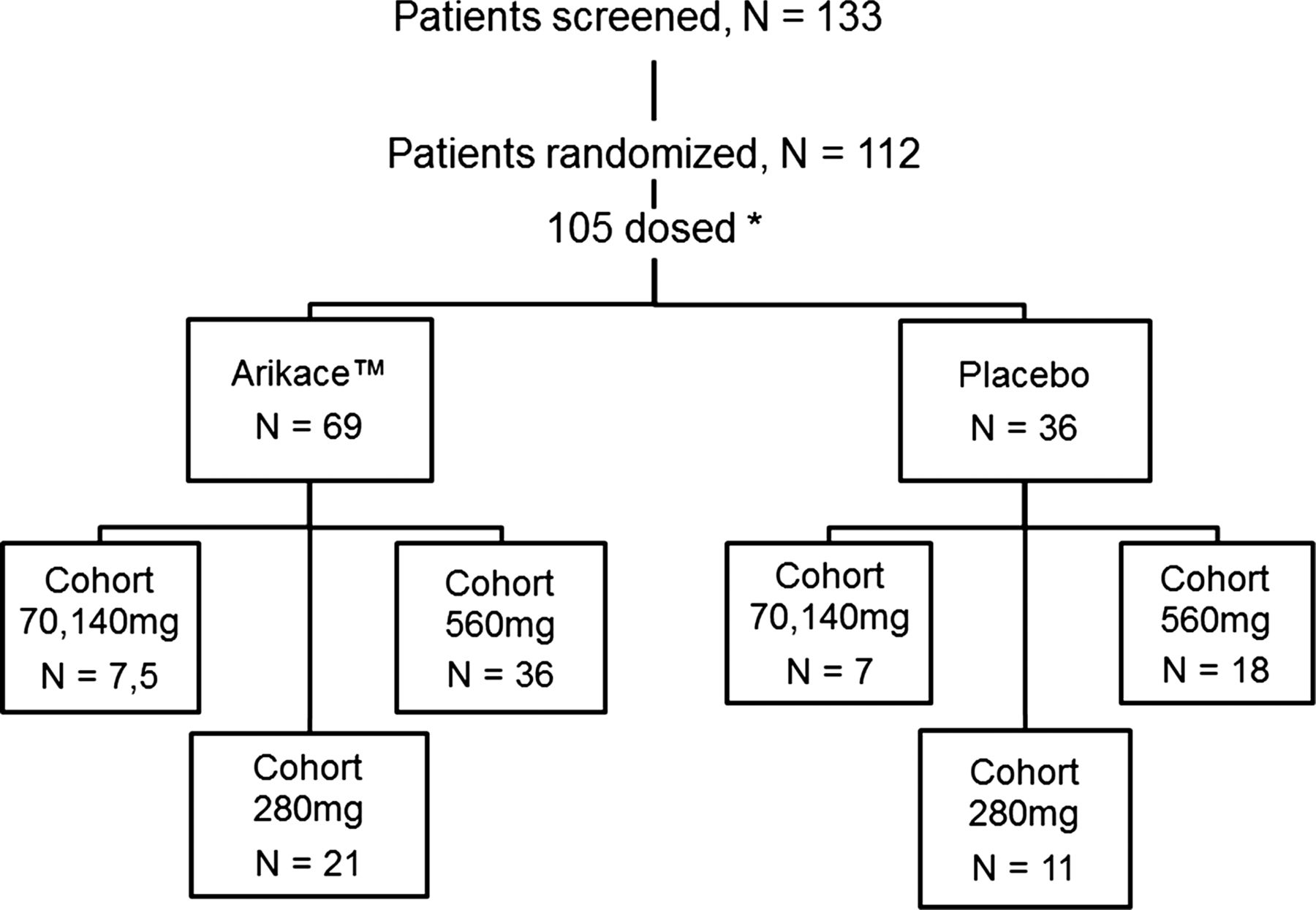
A total of 105 CF subjects meeting enrolment criteria were screened, randomised and dosed in one of four Arikace dose groups or placebo (figure 1, and enrolment segregated by US and European sites: see online supplementary figure S1). Subjects (across both studies) were randomised to receive 70 mg (n=7), 140 mg (n=5), 280 mg (n=21), 560 mg (n=36), or placebo (n=36). Baseline demographic characteristics (all subjects) are provided in table 1 (and for each study: see online supplementary tables S1 and S2). Age, gender, body mass index (BMI), lung function (FEV1% predicted) and chronic TIS use were generally similar, but with milder disease in the 140 mg dose group (higher FEV1 and BMI, US study, n=5), and younger age in the 280 mg cohort (16 years, European study, n=21). Mucoid P aeruginosa infection rates were similar in the European and US studies (85% and 89%, respectively), while chronic TIS use was lower in the European versus US subjects (defined as >three 28-day cycles in the preceding 12 months, 19% and 35%, respectively). The European cohort was younger (median age of 16.5 years (±6 years, SD) compared with 30.5 years (±8 years, SD) for the US cohort), but with similar lung function (median FEV1% predicted, 65.7% (±20%) for Europe versus 65.3% (±19%) for USA).
Demographic summary of all cystic fibrosis patients dosed in the European and US trials (mean (SD))

Patient enrolment across the two Arikace studies. In the European study, 75 patients were screened, and 66 were randomised. In the US study, 56 patients were screened, and 46 were randomised. *Across all randomised subjects, seven subjects withdrew consent prior to dosing. Data shown is for the 105 subjects dosed at least once with Arikace or placebo.
Safety and adverse event profile
Adherence and drug accountability was monitored by calculating the used and unused treatment vials returned at study visits. While an imperfect method to assess adherence, no difference was observed between the placebo and treatment groups (>90%). The overall frequency of AEs was similar across the Arikace dose groups and placebo, with no statistically significant differences. Acute tolerability was similar, with 2.8% of Arikace-treated subjects and 11.1% of placebos demonstrating a drop in FEV1 of ≥15% within 30 min of dosing. table 2 summarises AEs that occurred in ≥8% of Arikace-treated patients. The total number of subjects reporting at least one AE in the 560 mg Arikace group was 20 (55.6%) compared with 22 (61.1%) in the placebo group. The most frequent AEs were respiratory, occurring in 45% of all Arikace-treated versus 39% of placebo-treated subjects. The AE profile was generally similar, with more dysphonia reported in the high-dose Arikace group (8% vs 0%). Five subjects in the Arikace groups discontinued study drug (70 and 560 mg dose groups; due to respiratory events, dysphonia, laryngitis and tinnitus) compared with one placebo (respiratory event). AEs considered severe (grade 3) are listed in online supplementary table S3, and all related AEs are listed in online supplementary table S4 (all reported AEs are listed in online supplementary table S5). Additionally, eight SAEs occurred in the Arikace treatment groups (respiratory), including five pulmonary exacerbations with hospitalisation (four after Arikace dosing). Two placebo subjects were hospitalised (one for a pulmonary exacerbation, day 43; second subject was hospitalised twice (migraine, day 46 and elevated LFTs, day 79)). There were no other clinically significant changes in laboratory findings. Results from audiology testing (see online supplementary table S6) showed no differences between groups.
Adverse events (AE) occurring in ≥8% in Arikace-treated patients compared with placebo (European and US trials combined)
Spirometry
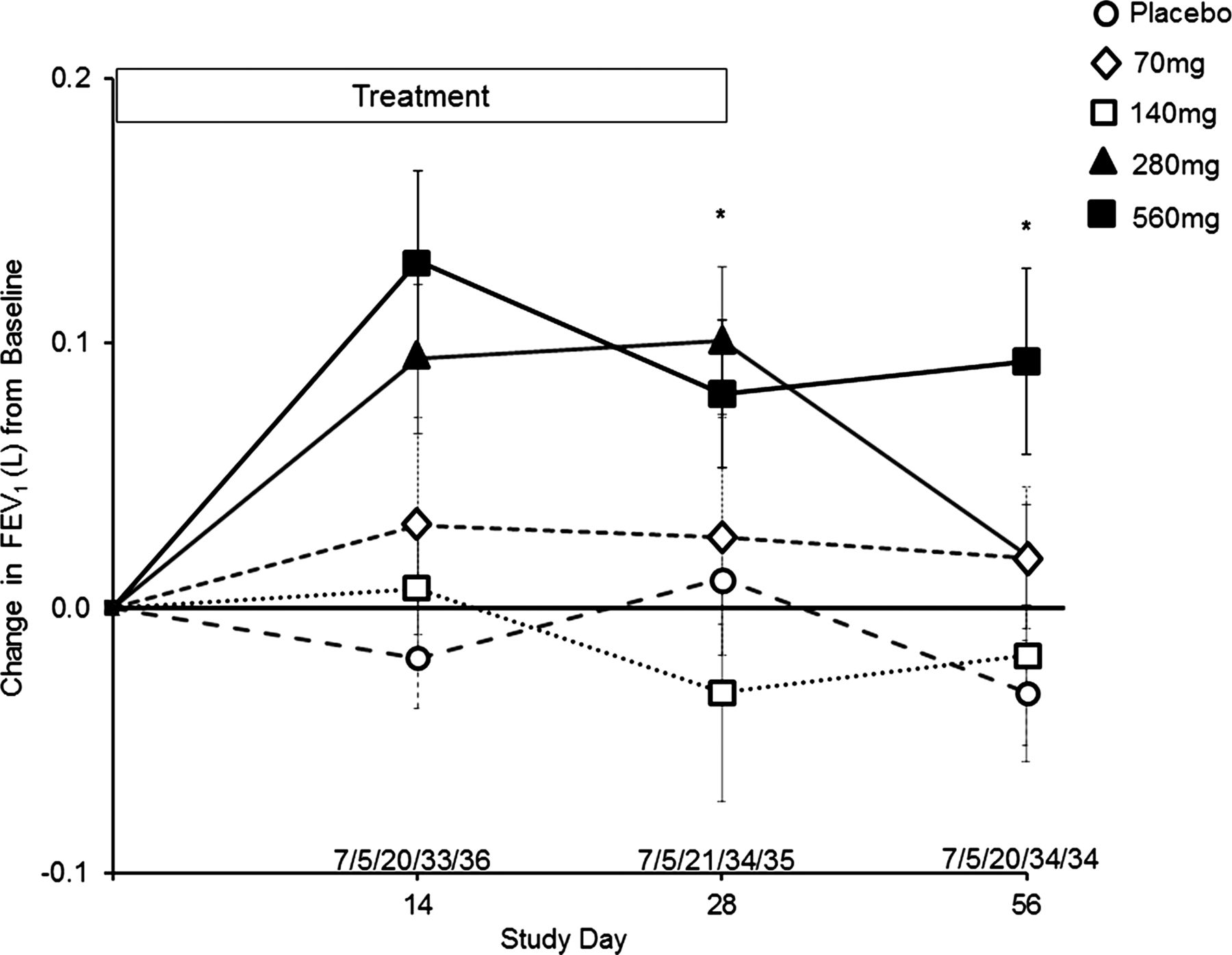
Pulmonary function data are summarised in figure 2. Due to the short duration of the placebo-controlled trial (28 days of dosing), data in the figure are presented as mean raw litre flows and SD (reduced risk of height measurement errors that can occur when reporting the FEV1% predicted). FEV1 in the placebo, 70 and 140 mg dose groups demonstrated no consistent trends over the 28-day treatment period. By contrast, the 280 mg and 560 mg dose groups demonstrated rapid, sustained and significant increases in FEV1 at days 14 and 28 compared with pretreatment values and placebo. In the 280 mg dose group, the change in FEV1 from baseline was higher at day 28 compared with placebo (0.101L±0.128 vs 0.011L±0.101; p=0.009), returning to pretreatment values by day 56 (28 days off study drug). In the 560 mg dose group, the change in FEV1 from baseline was higher at day 28 compared with placebo (0.081L±0.161 vs 0.011L ± 0.101; p=0.033), persisting through day 56 (0.093L±0.203 vs −0.032L±0.119; p=0.003). This corresponded to an % FEV1 predicted treatment effect at day 56 of 12.5%.
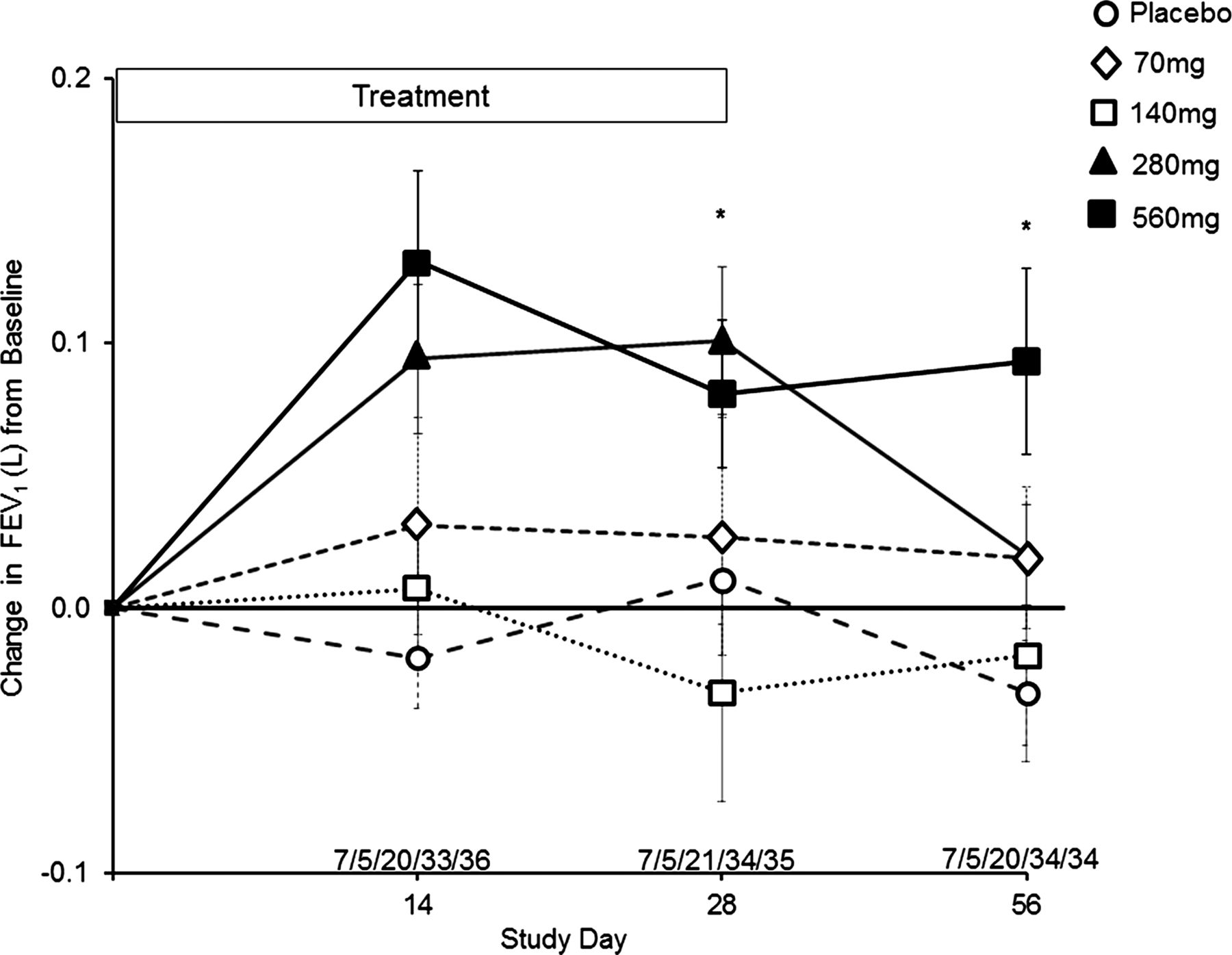
Change in FEV1 (L) from baseline through day 56. Filled squares, solid line=Arikace 560 mg, *p=0.033 at day 28, *p=0.003 at day 56 (compared with placebo). Filled triangles, solid line=Arikace 280 mg, *p=0.009 at day 28 (compared with placebo). Open squares, dashed line=Arikace 140 mg. Open diamonds, dashed line=Arikace 70 mg. Open circles, dashed line=placebo. The values above the abscissa are the number of subjects in each dose cohort providing data at each time point (70 mg/140 mg/280 mg/560 mg/placebo).
Microbiology
The mean baseline log10 P aeruginosa sputum density was 7.031 (±1.496) in the placebo group and 8.392 (±0.510), 8.000 (±1.047), 6.925 (±1.265), and 7.444 (±1.038) in the 70, 140, 280 and 560 mg dose groups, respectively. Rapid and sustained reduction of P aeruginosa sputum densities was noted for the 560 mg dose group compared with placebo (p=0.007) (figure 3). P aeruginosa sputum density remained reduced relative to baseline and placebo for the 560 mg dose group at day 35 (7 days off of study drug, p=0.021). Amikacin MIC for 50% of organisms (MIC50) and MIC for 90% of organisms (MIC90) values did not change over the 28-day treatment periods relative to placebo (data not shown).
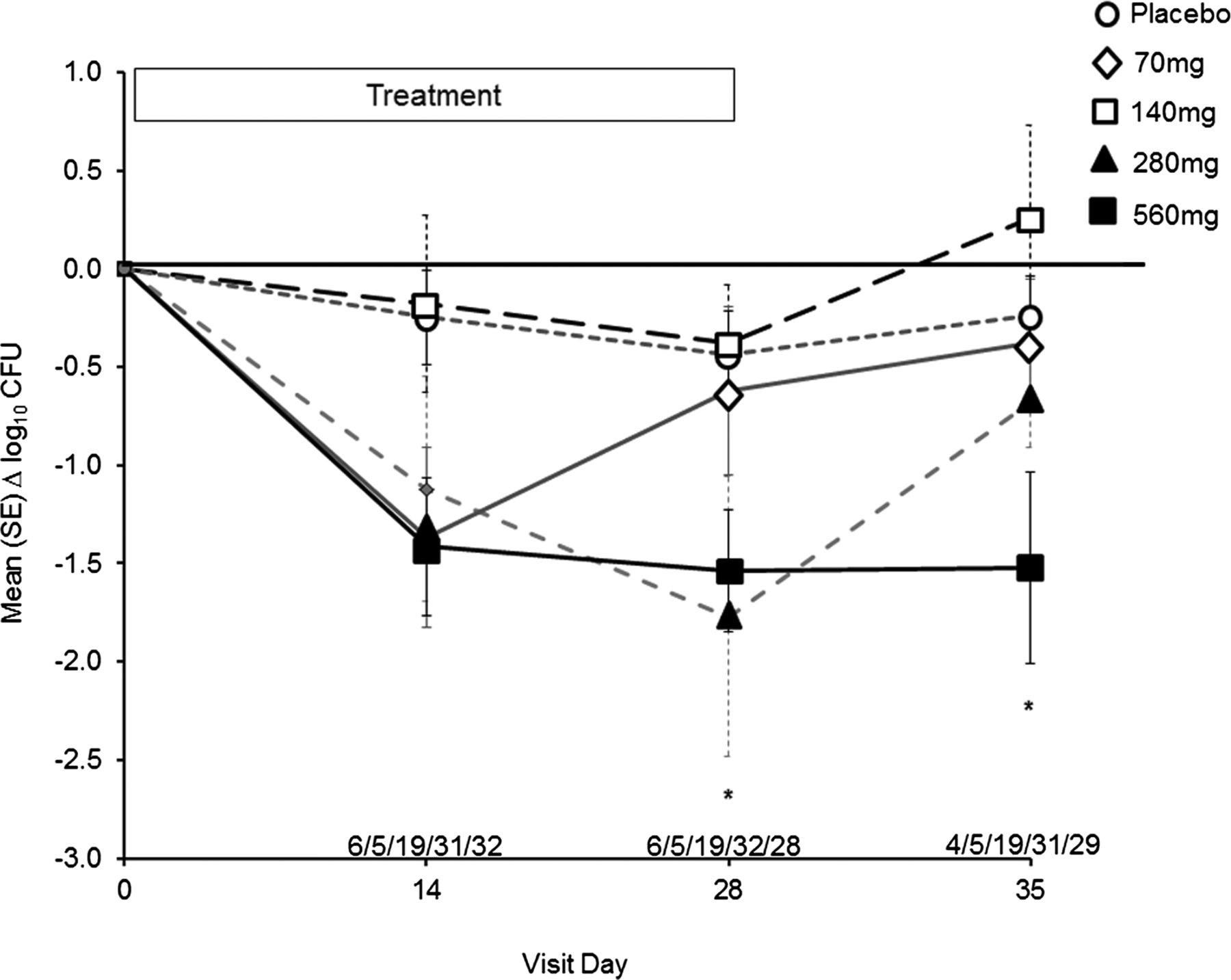
Change in sputum density of Pseudomonas aeruginosa (log10 CFU/g) from baseline through day 35. Filled squares, solid line=Arikace 560 mg, *p=0.007 at day 28, *p=0.021 at day 35. Filled triangles, solid line=Arikace 280 mg. Open squares, dashed line=Arikace 140 mg. Open diamonds, dashed line=Arikace 70 mg. Open circles, dashed line=placebo. The values above the abscissa are the number of subjects in each dose cohort providing data at each time point (70 mg/140 mg/280 mg/560 mg/placebo).
Patient-reported outcomes is included in the online supplementary figure S2.
Pharmacokinetic/pharmacodynamic results is included in the online supplement.
Open-label extension
A subgroup of subjects in the European trial (n=49) were enrolled into an open-label extension study to evaluate the safety, tolerability and efficacy of six repeat cycles of Arikace treatment (560 mg daily for 28 days) followed by 56 days off treatment. Baseline characteristics of this group are included in table 3, and changes in FEV1% predicted, P aeruginosa sputum CFUs/g, and the distribution of median MIC50 of Pseudomonas isolates are shown in figures 4, 5, and online supplementary figure S3, respectively. Repeat dosing with Arikace was well tolerated with four subjects discontinuing study drug over six cycles and 15 subjects experiencing SAEs (pulmonary exacerbations requiring treatment with antibiotics). Online supplementary table S7 summarises the AEs reported during the open-label extension. Of 49 subjects, 48 experienced at least one AE, with approximately one-third experiencing an SAE (none related to study drug). The majority of reported AEs (59%) were mild and most were classified as either infectious or respiratory. Of the 351 total AEs reported, 33 were categorised as possibly or probably related to study drug, and there were no deaths. FEV1% predicted (figure 4) demonstrated rapid and sustained increases for each treatment cycle, with an estimated mean increase of FEV1 (%) of 7.9% from baseline to end of the 28-day dosing period for the six cycles (p<0.0001), and a mean increase of 5.7% from baseline to end of day 84 (56 days post-treatment) across all six cycles (p=0.0001). P aeruginosa CFU/g were reduced across the treatment cycles (log10 CFUs of −0.53 at day 28 of the first treatment cycle (p=0.025)), with an estimated mean change in log10 CFUs of −0.60 from baseline over all measurements (p=0.003) across all six treatment cycles (figure 5). Median MIC50 values for P aeruginosa isolates demonstrated no significant change over all cycles (see online supplementary figure S3).
Demographic information of cystic fibrosis subjects enrolled in the open-label extension study evaluating repeated cycles of once daily 560 mg Arikace (mean (SD)—all from European sites)
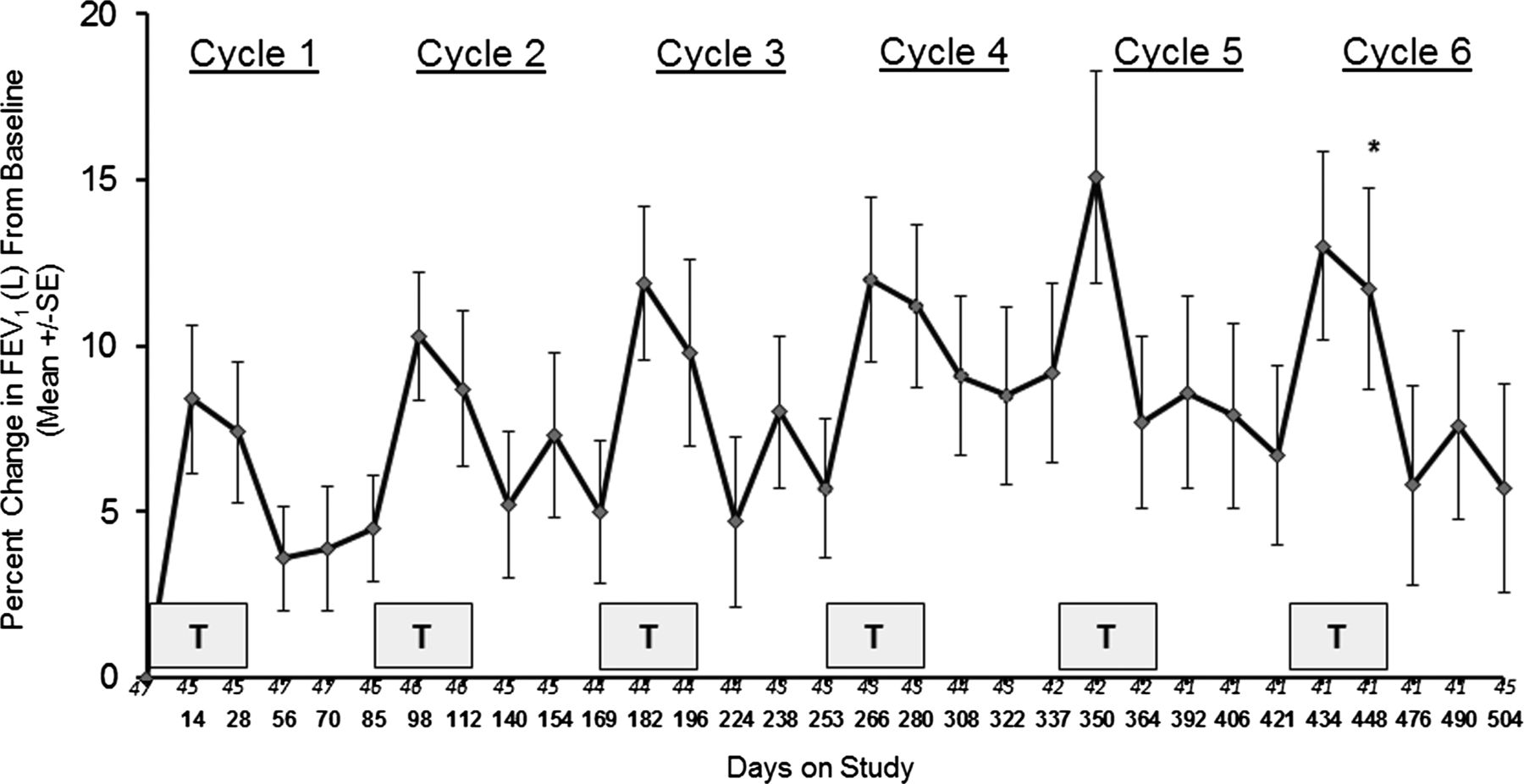
Change in FEV1 (% predicted) from baseline through cycle 6 of Arikace. Each cycle consisted of 28 days of once daily Arikace (560 mg) followed by 56 days off study drug. Each shaded box is a treatment cycle. Study days (every 2 weeks) are as shown on the abscissa, with the number of subjects at each time point as noted immediately above the study days. *p<0.0001 for FEV1 at end of treatment following six cycles compared with baseline; **p=0.0001 for FEV1 at 56 days post-treatment following six cycles compared with baseline.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in sputum density of Pseudomonas aeruginosa (log10 CFU/g) from baseline through cycle 6 of Arikace. Each set of three bars is the change in sputum P aeruginosa density compared with baseline (day 1 of cycle 1) for days 1 (white), 14 (gray), and 28 (black) of each respective Arikace cycle. *p=0.003 for change in CFU across all of the Arikace treatment cycles relative to baseline (cycle 1, day 1).
Discussion
Chronic P aeruginosa infection is a common problem in CF associated with accelerated loss of lung function, and is a predictor of morbidity and mortality. In the Phase II studies described, once-daily liposomal amikacin improved lung function, reduced the P aeruginosa sputum density, and improved patient-reported respiratory symptoms in CF patients over 28 days of treatment. Improvements in lung function persisted significantly above that of placebo-treated patients for 28 days post-treatment, and 56 days post-treatment over multiple cycles relative to baseline values. These prolonged benefits are novel and provide support for further evaluation of Arikace. Our findings contrast with those recently reported with other cycled, topical antibiotic preparations approved for the treatment of CF patients infected with P aeruginosa, where lung function returned towards pretreatment values off-cycle.9 ,11 ,14 ,15 Recent results of an open-label trial of nebulised cayston and TIS in CF patients with moderate lung disease reported that FEV1 changed minimally over three 28 days treatment cycles (+2.05% and −0.66% respectively, relative to baseline).16 Additionally, a recent phase III open-label study of colistimethate sodium dry powder in CF patients reported non-inferiority relative to TIS over three 28 day treatment cycles, with minimal change in lung function.13 Baseline lung function in these open-label studies was similar to ours (FEV1=49–59%), suggesting that this was not responsible for the differences observed across the trials.
Safety and tolerability of Arikace were supported by the AE and SAE profile, with similar rates of drug discontinuation across the Arikace and placebo subjects. There were no differences in audiologic or renal safety outcomes for the Arikace groups relative to placebo, which are relevant for the development of chronic nebulised aminoglycosides. The amikacin MIC50 of P aeruginosa isolates from the Arikace subjects (randomised and open-label extension) did not show appreciable increases over single or numerous treatment cycles. Although longer and larger trials of Arikace will be necessary to confirm our findings, the results provide reassurance for the design and execution of definitive trials.
Liposomal components of Arikace are found in lung surfactant, which is normally cleared rapidly by several lung cell types, (type II pneumocytes, Clara cells, and macrophages).22 Turnover rates in the adult human lung for DPPC and cholesterol are approximately 79 mg/h and 24 mg/day, respectively.23–25 Following a single 560 mg Arikace dose (via eFlow with 30% drug deposition), approximately 73 mg of DPPC (29 mg to lung periphery) and 36 mg of cholesterol (14 mg to lung periphery) are delivered to the lung. Repeated dosing over prolonged periods in animal models has little lasting effect on lung histology or pulmonary macrophage function.20 ,26 Together, these novel characteristics make Arikace unique among nebulised antibiotics.
The pharmacokinetics and pharmacodynamics of Arikace demonstrated high sputum-amikacin levels, low systemic levels, and dose-dependent relationships between Arikace and both FEV1 and FEF25−75% improvements. These pharmacokinetic data (see online supplementary table S8) support prolonged deposition of amikacin in the lung, with stable airway clearance, but increasing systemic clearance of amikacin with increasing Arikace exposure. This is likely a product of enhanced lung deposition, and the safety of this systemic exposure will need to be considered in future study designs. The data from the open-label extension (figure 4 and see online supplementary table S7) support long-term tolerance over repeated cycles, but this will also require confirmation in future studies. The consistent reductions in P aeruginosa density (figure 5), coupled with prolonged improvements in lung function and respiratory symptoms over repeated cycles (figure 4), support a prolonged antimicrobial effect. The MIC50 for P aeruginosa isolates was increased in later cycles, but did not demonstrate a clear trend over the entire open-label study, or within individual cycles (see online supplementary figure S3). Monitoring of aminoglycoside susceptibility patterns will be needed in future studies of Arikace, including resistance to amikacin and tobramycin.
Important limitations in the study include the small lower dose Arikace cohorts, the younger age and relatively more severe lung disease in the European cohorts, and the relatively low treatment with chronic inhaled tobramycin (likely reflective of strict definition criteria). Despite these limitations, our data provide evidence for the short-term safety, tolerability and efficacy of nebulised Arikace in CF patients chronically infected with P aeruginosa. The current data supports future studies with a once-daily 560 mg dose using a rapid delivery nebuliser, with prolonged off-treatment cycles. Higher doses, or more frequent dosing, may be appropriate for other conditions (eg, non-tuberculous mycobacteria infection), and are under investigation (NCT01315236). These dosing features may potentially enhance adherence and minimise chronic drug exposure, thereby reducing the risk of long-term side effects. These benefits may signal a new approach for the management of chronic pulmonary infections utilising antibiotics combined with neutral liposomes.
Acknowledgments
Pharmacokinetic/Pharmacodynamic analyses The authors gratefully acknowledge Olanrewaju O Okusanya from the Institute for Clinical Pharmacodynamics, Latham, NY, for providing the analyses for this manuscript. The authors are also grateful to J Denise Wetzel, CCHMC Medical Writer, for critical review of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
Footnotes
JPC and LD contributed equally.
The Altera Nebulizer System is a registered trademark of PARI Respiratory Equipment (2943 Oak Lake Blvd, Midlothian, VA 23112, USA).
-
Collaborators Site PIs of the Arikace Study Group: US Study PIs: Dr J Clancy—Cincinnati Children's Hospital Medical Center, Cincinnati, OH. Dr R Young—University of Alabama at Birmingham, Birmingham, AL. Dr R Ahrens—University of Iowa, Iowa City, IA. Dr M Aitken—University of Washington, Seattle, WA. Dr J Billings—University of Minnesota, Minneapolis, MN. Dr A Faro—Washington University School of Medicine, St Louis, MO. Dr C Goss—University of Washington, Seattle, WA. Dr D Layish—Central Florida Pulmonary Group, Orlando, FL. Dr N Lechtzin—Johns Hopkins Pulmonary and Critical Care Medicine, Baltimore, MD. Dr M Light—University of Miami, Miami, FL. Dr S Miller—University of Mississippi, Jackson, MS. Dr S Nasr—University of Michigan Health System, Ann Arbor, MI. Dr J Nick—National Jewish Medical and Research Center, Denver, CO. Dr RC Rubenstein—The Children's Hospital of Philadelphia and Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA. Dr A Sannuti—Indiana University School of Medicine, Indianapolis, IN. Dr G Sawicki—Children's Hospital Boston, Harvard, Boston, MA. Dr J Taylor-Cousar—University of New Mexico, Albuquerque, NM. Dr B Trapnell—Cincinnati Children's Hospital Medical Center, Cincinnati, OH. Dr J Wallace—Sanford Clinic—Clinical Research, Sioux Falls, SD: European Study PIs: Professor P Minic—Institute for Mother and Child Healthcare, Belgrade, Serbia, Professor S Fustik—University Clinical Centre, Skopje, Macedonia. Dr E Solyom—BAZ County Hospital, Miskolc, Hungary, Professor H Mazurek—Institute of Tuberculosis and Lung Diseases, Rabka Zdrój, Poland, Professor Y Antipkin—Institute of Pediatrics, Obstetrics and Gynecology AMS, Kyiv, Ukraine. Dr A Feketeova—Children University Hospital Košice, Košice, Slovak Republic, Professor A Senatorova—Kharkiv Regional Children Clinical Hospital, Kharkiv, Ukraine. Dr E Csiszer—Korányi TBC és Pulmonológiai Intézet, Budapest, Hungary, Professor V Kostromina—Yanovsky Institute of Phtisiology and Pulmonology, Kiev, Ukraine. Dr B Takac—Children University Hospital (DFNsP), Banská Bystrica, Slovak Republic. Dr R Ujhelyi—Heim Pál Children Hospital, Budapest, Hungary. Dr A Sovtić—Institute for Mother and Child Healthcare, Belgrade, Serbia; Laboratory Support: Anne Marie Buccat—Laboratory Supervisor, Seattle Children's Hospital, Seattle, WA, Catherine Doherty—CF Group, University of Edinburgh Medical School, Edinburgh, UK.
-
Contributors US study design, execution, and analysis: JPC, MK, JB, CG, JL, ALQ, RCC, KRY, LS, JB, BR, RG. European study design, execution, and analysis: LD, SF, PM, JRWG, JL, ALQ, RG. Interaction with regulatory authorities: JPC, MK, CG, JL, BR, ALQ, RG. Manuscript writing and review: JPC, LD, MK, JB, SF, CG, JL, PM, ALQ, RCC, KRY, LS, JB, BR, JRWG and RG.
-
Funding Grant support: This work was supported in part by a grant received by Insmed through a Trial Funding Agreement with the Cystic Fibrosis Foundation (CFF); NCT00558844, NCT00777296. JPC was supported by CFF grant CLANCY09YO. The Children's Hospital of Philadelphia and University of Pennsylvania CTRC were supported for this work by UL1RR024134 (NCRR) and UL1TR000003 (NCATS). The Seattle Children's TDN Center for CF Microbiology was supported for this work by CFF grant BURNS03Y2. LD received support for this work as a senior clinical research fellow of the FWO Flanders, Belgium. BWR is supported by CFF grant RAMSEY03YO. CHD: CFRSD – NIH/NCRR UL1 RR025014-03S5, P30 DK089507, and CFF grants PATRIC11A0 and GOSS05A0.
-
Competing interests None.
-
Ethics approval Local institutional review boards and ethics boards.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Questions regarding unpublished data from this trial should be directed to Insmed Corporation.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Linked Articles
- Airwaves