Article Text

Abstract
Background Cigarette smoking is an important risk factor for the development of cardiovascular disease, yet the pathways through which this may operate are poorly understood. Therefore, the mechanism underlying cigarette smoke (CS)-induced arterial endothelial dysfunction and the potential link with fractalkine/CX3CL1 upregulation were investigated.
Methods and results Stimulation of human arterial umbilical endothelial cells (HUAECs) with pathophysiological concentrations of CS extract (1% CSE) increased CX3CL1 expression. Neutralisation of CX3CL1 activity under dynamic flow conditions significantly inhibited CSE-induced mononuclear cell adhesion to HUAECs (67%). The use of small interfering RNA (siRNA) revealed that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 5 (Nox5) but not Nox2 or Nox4 is the main NADPH isoform involved in CSE-induced CX3CL1 upregulation and mononuclear cell arrest. Knock down of HUAEC tumour necrosis factor α expression with siRNA or pharmacological inhibition of p38 mitogen-activated protein kinase and nuclear factor κB also abolished these responses. Interestingly, circulating monocytes and lymphocytes from patients with chronic obstructive pulmonary disease (COPD) (n=29) versus age-matched controls (n=23) showed CX3CR1overexpression. Furthermore, CX3CL1 neutralisation dramatically diminished their enhanced adhesiveness to CSE-stimulated HUAECs. Finally, when animals were exposed for 3 days to CS, a mild inflammatory response in the lung was observed which was accompanied by enhanced CX3CL1 expression in the cremasteric arterioles, an organ distant from the lung. CS exposure resulted in increased leukocyte–arteriolar endothelial cell adhesion which was significantly reduced (51%) in animals lacking CX3CL1 receptor (CX3CR1).
Conclusions These results suggest that CS induces functional CX3CL1 expression in arterial endothelium and leukocytes from patients with COPD show increased CX3CL1-dependent adhesiveness. Therefore, targeting the CX3CL1/CX3CR1 axis might prevent COPD-associated cardiovascular disorders.
- COPD Mechanisms
- COPD Pathology
- COPD Pharmacology
- Oxidative Stress
- Tobacco and the lung
Statistics from Altmetric.com
Key messages
What is the key question?
-
How does cigarette smoke (CS) induce arterial endothelial dysfunction and leukocyte recruitment?
What is the bottom line?
-
Fractalkine/CX3CL1 is upregulated in the arterial endothelium after stimulation with CS extract, increasing the endothelial adhesiveness for CX3CL1 receptor (CX3CR1)-expressing cells. Blockade of the CX3CL1/CX3CR1 axis dramatically reduced the arterial adhesion of mononuclear leukocytes from patients with chronic obstructive pulmonary disease (COPD) to CS extract-stimulated endothelium.
Why read on?
-
This is the first report that has systematically characterised the underlying mechanisms involved in CS-induced arterial endothelial dysfunction. We have provided evidence that CX3CL1 upregulation is a critical molecule in CS-induced mononuclear leukocyte recruitment. Therefore, CX3CL1 and CX3CR1 may be considered as potential drug targets for the prevention and treatment of COPD-associated cardiovascular disorders.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterised by a progressive and largely irreversible decrement in lung function associated with an abnormal chronic inflammatory response of the lungs to noxious particles and gases, mostly from cigarette smoke (CS).1 In addition to the pulmonary features of COPD, several systemic effects have been recognised, such as skeletal muscle dysfunction, cardiovascular disease, osteoporosis and diabetes.1 Epidemiological studies demonstrate that smoking is a significant risk factor for heart disease, including aneurysm formation and rupture, stroke and atherosclerosis,2 which is one of the leading causes of morbidity and mortality in Western countries.3 One of the earliest stages of atherogenesis is endothelial dysfunction, which leads to a proinflammatory and prothrombotic phenotype of the endothelium4 and thus provokes the attachment and the subsequent migration of leukocytes. In this context, vascular dysfunction in smokers has been widely described.5 ,6
Adhesive interactions between leukocytes and arterial endothelium precede leukocyte infiltration to the subendothelial space.7 The migration of leukocytes from the blood to sites of extravascular injury is mediated through a sequential cascade of leukocyte–endothelial cell adhesive interactions which involve an array of cell adhesion molecules (CAMs) present on leukocytes and endothelial cells.7 In addition to CAMs, chemokines have the potential to recruit specific cell types and are involved in the regulation of leukocyte trafficking.8 Fractalkine (CX3CL1) is the unique member of the CX3C subfamily and is expressed in a soluble and membrane-bound form on the surface of inflamed endothelium. As a full-length transmembrane protein, CX3CL1 acts as an adhesion molecule.9 Cleavage of the CX3CL1 mucin stalk close to the junction with the transmembrane domain produces a soluble form of CX3CL1 that is a potent chemoattractant for monocytes and T cells but not for neutrophils.10 It interacts with leukocytes expressing its receptor CX3CR1. The ability of fractalkine to attract and arrest blood monocytes and lymphocytes, and its presence in vascular wall cells makes it an attractive candidate for playing a pivotal role in atherosclerotic lesion formation. Indeed, independent studies with CX3CR1−/− apolipoprotein E−/− or CX3CR1−/− LDLr−/− mice have associated a substantial decrease in macrophage infiltration within the arterial wall with a marked reduction in atherosclerosis development, suggesting a relevant role for the CX3CL1/CX3CR1 axis.11 ,12
The mechanisms by which CS promotes the development of a proinflammatory environment in the vessel wall are not fully understood and experimental data evaluating the impact of CS within the cardiovascular system using in vitro and in vivo approaches are scarce. Therefore, in this study we sought to determine whether CS induces functional CX3CL1 expression in organs distant from the lung and the underlying mechanisms involved in these responses. In vitro experiments were carried out in primary cultures of human arterial endothelial cells stimulated with CS extract (CSE). Additionally, to explore the clinical consequences of our findings, CX3CR1 receptor expression in different circulating leukocyte subsets from patients with chronic obstructive pulmonary disease was analysed and their CX3CR1-dependent adhesiveness to human umbilical arterial endothelial cells (HUAECs) evaluated. Finally, intravital microscopy within the murine cremasteric microcirculation was used to determine leukocyte–endothelial cell interactions induced by CS exposure.
Methods
Human in vitro studies
All investigation with human samples carried out in the present study conforms with the principles outlined in the Declaration of Helsinki and was approved by the institutional ethics committee at the University Clinic Hospital of Valencia, Spain. Written informed consent was obtained from all volunteers.
CSE preparation
Cigarettes were obtained from the Kentucky Tobacco Research and Development Center at the University of Kentucky. CSE was prepared as described previously.13 Further details are described in the online data supplement.
Reverse transcriptase PCR
Total RNA was isolated from cultured HUAECs by using Trizol Isolation Reagent. Details are found in the online data supplement.
Flow cytometry
After treatments, endothelial cells were detached from culture flasks by treatment with ice-cold phosphate-buffered saline containing 0.05% NaN3 and 0.2% bovine serum albumin, subsequent scraping and centrifugation. Details are described in the online data supplement.
Leukocyte–endothelial cell interactions under flow conditions
HUAECs were grown to confluence and subjected to different treatments. Further details are found in the online data supplement.
Immunofluorescence
CX3CL1 expression was visualised in HUAECs by indirect immunofluorescence. Details are described in the online data supplement.
Western blot
CX3CL1 expression was also determined by immunoblotting. Further details are provided in the online data supplement.
Transfection of tumour necrosis factor α, Nox2, Nox4 or Nox5 small interfering RNA
Endothelial cell gene silencing was performed using either control or tumour necrosis factor α (TNFα), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox2), Nox4 or Nox5-specific small interfering RNA. Details are described in the online data supplement.
Experimental protocols
Details of all the experimental protocols followed in this study are provided in the online data supplement.
Studies in patients with COPD and age-matched controls
Patients’ details, the procedure for determining CX3CR1 expression on circulating leukocytes, their adhesiveness to CSE-stimulated HUAECs and plasma CX3CL1 levels can be found in the online data supplement.
In vivo animal studies
CS exposure
Exposure of mice to CS was carried out using a modified method previously described.14 Details of the procedure are found in the online data supplement.
Intravital microscopy
Intravital microscopy was carried out in the mouse cremasteric microcirculation. Details of the technique are provided in the online data supplement.
Histology and immunohistochemistry
Immunofluorescence studies were performed in the cremasteric microvasculature. Details of the procedure are found in the online data supplement.
Materials
All materials used are listed in the online data supplement.
Statistical analysis
The statistical analyses used are provided in the online data supplement.
Results
CSE induces functional CX3CL1 expression in HUAECs
HUAECs were incubated with different CSE concentrations (0.1%–3%) or TNFα (20 ng/ml) for 1 or 4 h. Reverse transcriptase PCR revealed that after 1 h of incubation with CSE no changes in CX3CL1 mRNA expression were detected whereas TNFα provoked a clear enhancement (figure 1A). When cells were stimulated with CSE for 4 h, a concentration-dependent increase in CX3CL1 mRNA expression was observed (figure 1B). Based on these results, the concentration of 1% CSE was used for the reminder experiments. This concentration approximately corresponds to exposures associated with smoking 1.5 packs per day as previously estimated15 and it is consistent with the amount smoked by the patients with COPD used in this study. In addition, it did not cause cytotoxicity to endothelial cells as found with the 3% CSE concentration (assessed by MTT and lactate dehydrogenase release assays, data not shown).
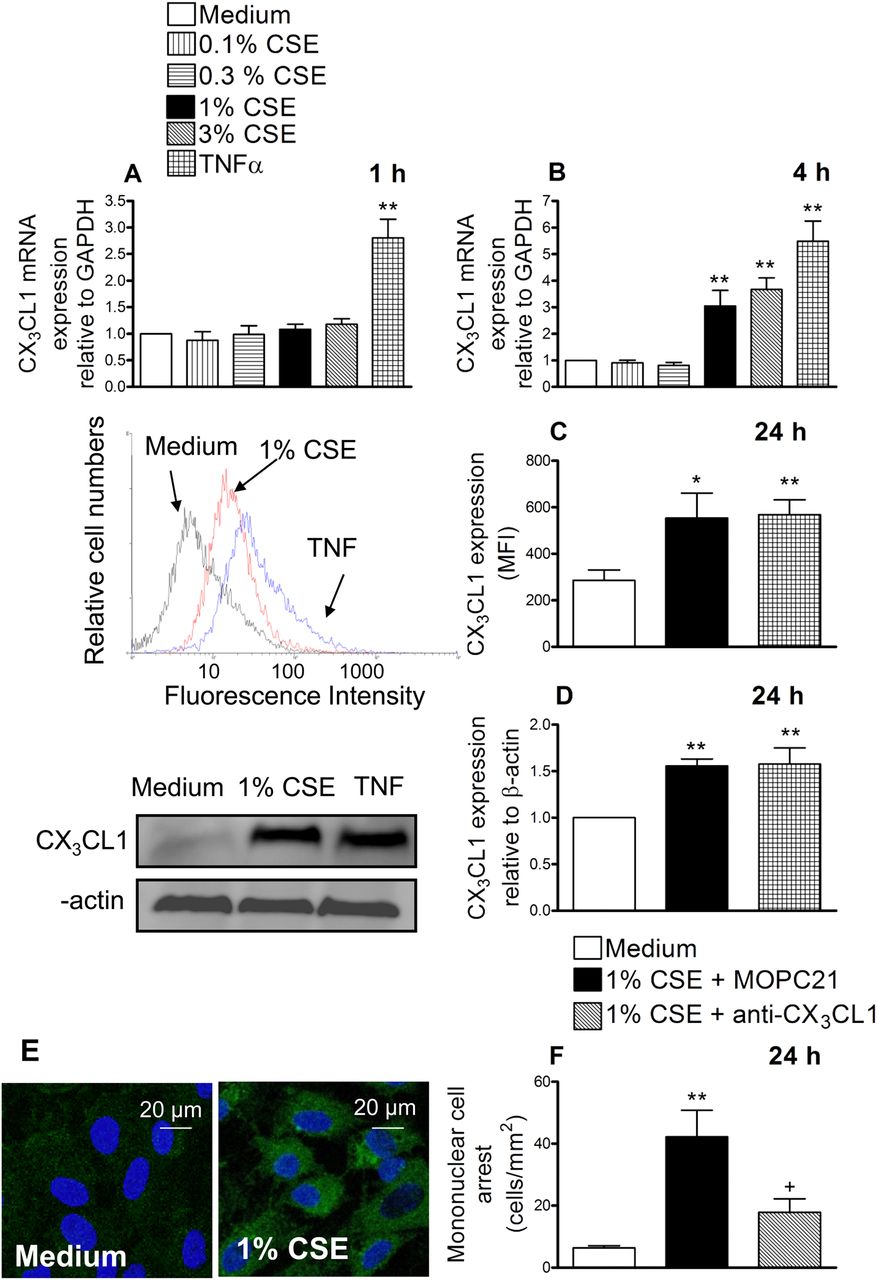
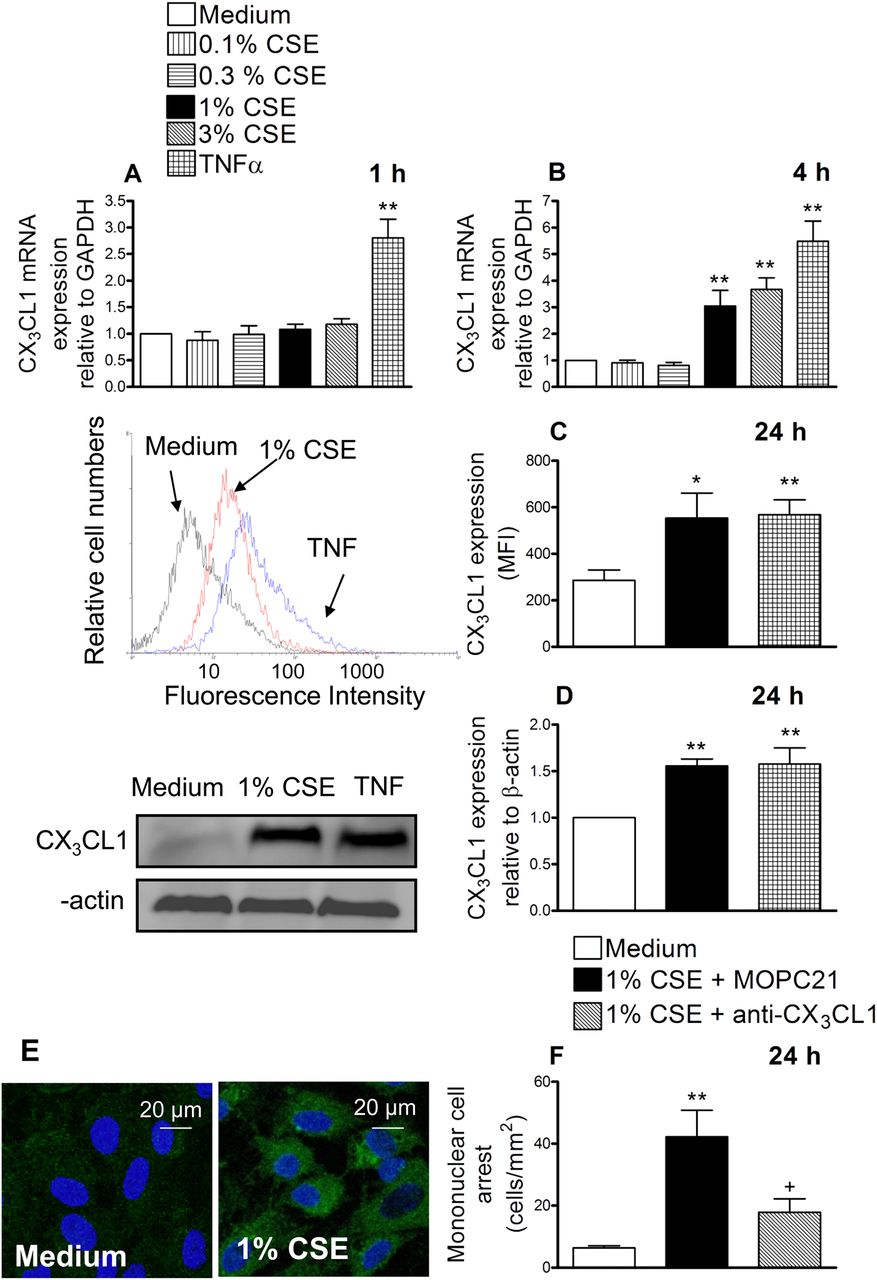
Cigarette smoke extract (CSE) induces CX3CL1 mRNA and protein expression in human arterial umbilical endothelial cells (HUAECs) (A–E). A neutralising antibody against CX3CL1 function inhibited the recruitment of mononuclear leukocytes to CSE-stimulated HUAECs (F). HUAECs were stimulated with CSE (0.1–3%) or tumour necrosis factor α (TNF α) (20 ng/ml) for 1, 4 or 24 h. Relative quantification of mRNA levels for CX3CL1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was determined by using reverse transcriptase PCR by the comparative Ct method (ΔΔCt method). Columns show the fold increase in expression of CX3CL1 mRNA, relative to control GAPDH values, calculated as mean±SEM of the 2−ΔΔCt values (n=3–4 independent experiments). Protein expression was determined by flow cytometry and expressed as mean fluorescence intensity (MFI). Representative histograms are also shown (mean±SEM of n=7 independent experiments). Following a similar protocol, CX3CL1 expression was also determined by western blotting. Results (mean±SEM of n=5–6 independent experiments) are expressed as fold increase in CX3CL1:β actin. Representative gels are shown above.*p<0.05 or **p<0.01 relative to values in the medium group. CX3CL1 upregulation was visualised in non-permeabilised HUAECs by immunofluorescence (green). Nuclei were counterstained with 4'6-diamidino-2-phenylindole (DAPI). Results are representative of n=5 independent experiments. Endothelial cells were stimulated with 1% CSE for 24 h. Some cells were incubated with a neutralising antibody against CX3CL1 function (5 μg/ml) or with an irrelevant isotype-matched monoclonal antibody (MOPC21, 5 μg/ml). Then human mononuclear cells (1×106 cells/ml) were perfused over the monolayers for 5 min at 0.5 dyn/cm2 and leukocyte accumulation quantified. Results are the mean±SEM of n=7 independent experiments. *p<0.05 or **p<0.01 relative to values in the medium group; +p<0.05 relative to 1% CSE group.
Therefore, we next evaluated the effect of CSE at the protein level. Endothelial cells were stimulated with 1% CSE for 24 h and analysed by flow cytometry. CX3CL1 expression was detected in CSE-stimulated HUAECs (figure 1C). We further confirmed these observations by western blot (figure 1D) and immunofluorescence (figure 1E). To investigate the functional role of CSE-induced CX3CL1 expression, freshly isolated human mononuclear cells were perfused across a monolayer of HUAECs stimulated with 1% CSE for 24 h. As illustrated in figure 1F, significant increases in mononuclear cell arrest were observed in CSE-stimulated cells. Interestingly, neutralisation of CX3CL1 activity on the endothelial cell surface resulted in a significant reduction in CSE-induced mononuclear cell adhesion to HUAECs (67% inhibition), suggesting a potential role for this chemokine in the arterial mononuclear leukocyte recruitment associated with CS.
Nox5 but not Nox2 or Nox4 siRNA inhibits CSE-induced CX3CL1 expression and mononuclear cell arrest in HUAECs
CS-mediated oxidative stress is implicated in endothelial dysfunction and water-soluble components of CS smoke can increase reactive oxygen species (ROS) generation in endothelial cells.16 Potential sources of superoxide anion in the vasculature include the activation of Noxs and xanthine oxidase (XO). Figure 2 illustrates that the inhibition of Nox with apocynin reduced CSE-induced CX3CL1 expression and mononuclear cell adhesion by 81% and 93% respectively. XO inhibition with allopurinol did not exert any significant inhibition.

Cigarette smoke extract (CSE)-induced CX3CL1 expression (A) and mononuclear cell arrest (B) is inhibited by apocynin but not by allopurinol. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 5 (Nox5) but not Nox2 or Nox4 small interfering RNA (siRNA) inhibits CSE-induced CX3CL1 expression and mononuclear cell arrest in human arterial umbilical endothelial cells (HUAECs) (C–H). HUAECs were incubated with apocynin (30 μM) or allopurinol (100 μM) for 1 h and then stimulated with 1% CSE for 24 h. CX3CL1 expression was determined by flow cytometry. Results (mean±SEM of n=6–7 independent experiments) are expressed as mean fluorescence intensity (MFI) (A). Human mononuclear cells (1×106 cells/litre) were perfused over the monolayers for 5 min at 0.5 dyn/cm2 and leukocyte accumulation quantified (B). Results are the mean±SEM of n=6 independent experiments. **p<0.01 relative to values in the medium group; +p<0.05 relative to 1% CSE group. Endothelial cells were transfected with Nox2, Nox4 or Nox5 siRNA or control siRNA. 48 h post-transfection cells were stimulated with 1% CSE for 24 h. CX3CL1 expression was determined by flow cytometry. Results (mean±SEM of n=5–7 independent experiments) are expressed as MFI (C, E and G). *p<0.05 or **p<0.01 relative to values in the medium group; +p<0.05 relative to 1% CSE group in control siRNA transfected cells. Human mononuclear cells (1×106 cells/ml) were perfused over the monolayers for 5 min at 0.5 dyn/cm2 and leukocyte accumulation quantified (D, F and H). Results are the mean±SEM of n=5–7 independent experiments. *p<0.05 or **p<0.01 relative to values in the medium group; +p<0.05 relative to 1% CSE group in control siRNA transfected cells. XO, xanthine oxidase.
Vascular Noxs are expressed in a cell-specific manner, with endothelial cells expressing mainly Nox2, Nox4 and Nox5.17 ,18 To determine the Nox isoform implicated in CSE-induced responses, we used a siRNA approach for knocking down Nox2, Nox4 or Nox5 in HUAECs. Significant reductions in Nox2, Nox4 and Nox5 mRNA (71–80%) and CSE-induced protein expression (60–68%) were evident after 48 h of incubation with their respective siRNA (see Figure I in online data supplement). Notably, whereas downregulation of Nox2 or Nox 4 had no impact on CSE-induced CX3CL1 expression and mononuclear cell adhesion to HUAECs, Nox 5 silencing dramatically inhibited these responses (figure 2G–H).
TNFα siRNA inhibits CSE-induced CX3CL1 expression and mononuclear cell arrest in HUAECs
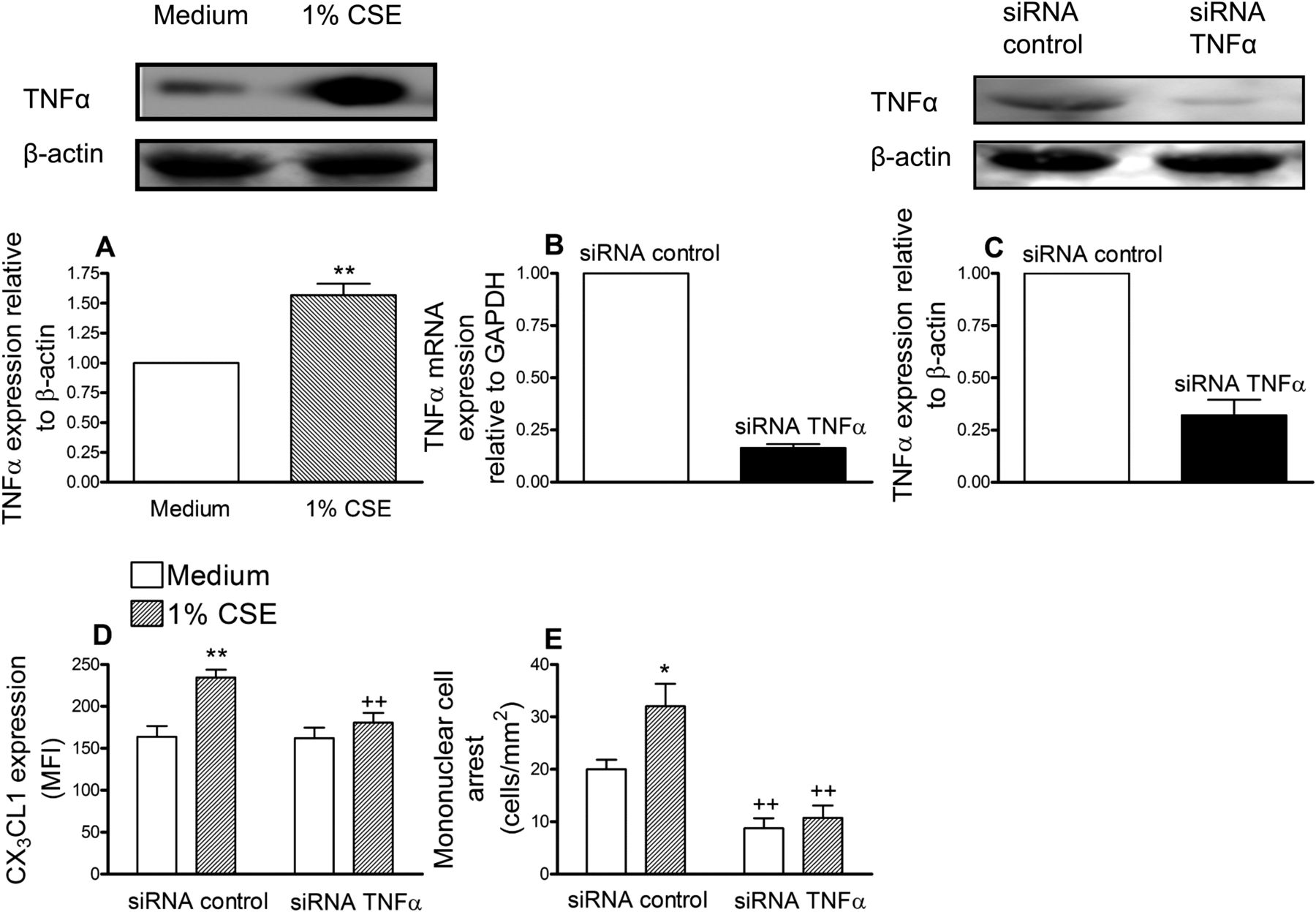
Increased serum levels of TNFα have been detected in healthy smokers119 and TNFα is one of the main inducers of CX3CL1 expression.9 We first evaluated the effect of CSE on TNFα expression in HUAECs and increased expression was encountered after 24 h of stimulation (figure 3A). To suppress TNFα expression, again a siRNA approach was used. HUAECs showed a >80% reduction in TNFα mRNA and >70% decrease in the intracellular cytokine compared with control siRNA-treated cells (figure 3B and C). CSE extract significantly increased the expression of CX3CL1 and caused mononuclear cell recruitment in control siRNA-transfected cells (figure 3D and E). However, in TNFα-deficient HUAECs, CSE-induced responses were significantly reduced (figure 3D and E).

Cigarette smoke extract (CSE) induces tumour necrosis factor α (TNFα) increased expression in human arterial umbilical endothelial cells (HUAECs) (A). TNFα small interfering RNA (siRNA) inhibits CSE-induced CX3CL1 expression (D) and mononuclear cell arrest (E) in HUAECs. HUAECs were stimulated with 1% CSE for 24 h. TNFα expression was determined by western blotting. Results (mean±SEM of at least four independent experiments) are expressed as fold increase of the TNFα:β actin. Representative gels are shown above. **p<0.01 relative to values in the medium group. Endothelial cells were transfected with TNFα siRNA or control siRNA. Knockdown of TNFα in HUAECs was determined by reverse transcriptase PCR (B) and western blotting (C). 48 h post transfection, cells were stimulated with 1% CSE for 24 h. CX3CL1 expression was determined by flow cytometry. Results (mean±SEM of n=6 independent experiments) are expressed as mean fluorescence intensity (MFI) (D). Human mononuclear cells (1×106 cells/ml) were perfused over the monolayers for 5 min at 0.5 dyn/cm2 and leukocyte accumulation quantified (E). Results are the mean±SEM of n=7–8 independent experiments. *p<0.05 or **p<0.01 relative to values in the medium group; ++p<0.01 relative to their respective group in control siRNA transfected cells.
P38 mitogen-activated protein kinase and nuclear factor κB are involved in CSE-induced CX3CL1 expression and mononuclear cell arrest in HUAECs
Generation of oxidants by CS appears to be the primary stimulus for activation of mitogen-activated protein kinase (MAPK) cascades at least in the lung epithelium.20 In this study, CSE stimulation caused a rapid phosphorylation of p38 MAPK and the p65 subunit of nuclear factor κB (NFκB) (figure 4A and B). Consequently, CSE-induced CX3CL1 expression and mononuclear cell adhesion were attenuated by pretreatment of the endothelial cells with the inhibitors of p38 MAPK or NFκB (figure 4C and D).

Cigarette smoke extract (CSE) induces p38 mitogen-activated protein kinase (MAPK) (A) and nuclear factor κB (NFκB) (B) activation in human arterial umbilical endothelial cells (HUAECs). CSE-induced CX3CL1 expression (C) and mononuclear cell arrest (D) in HUAECs is reduced by a p38 MAPK or a NFκB inhibitor. Cells were stimulated for 30 min or 1 h with 1% CSE. p38 MAPK and NFκB activation was determined by flow cytometry. Results (mean±SEM of at least six independent experiments) are expressed as mean fluorescence intensity (MFI) (A and B). Representative histograms are also shown. *p<0.05 or **p<0.01 relative to values in the control group. HUAECs were incubated with SB202130 (20 μM) or MOL294 (2.5 μM) for 1 h and then stimulated with 1% CSE for 24 h. CX3CL1 expression was determined by flow cytometry. Results (mean±SEM of n=6–7 independent experiments) are expressed as MFI (C). Human mononuclear cells (1×106 cells/ml) were perfused over the monolayers for 5 min at 0.5 dyn/cm2 and leukocyte accumulation quantified (D). Results are the mean±SEM of n=6–7 independent experiments. **p<0.01 relative to values in the medium group; +p<0.05 or ++p<0.01 relative to 1% CSE group.
Circulating mononuclear cells from patients with COPD show increased fractalkine receptor expression (CX3CR1) and adhesiveness to CSE-stimulated HUAECs compared with those from healthy controls
We next determined CX3CR1 expression on circulating monocytes and lymphocytes from patients with COPD and age-matched controls. As illustrated in figure 5A–D, increased and significant differences in CX3CR1 expression and the percentage of circulating leukocytes expressing it were detected in monocytes and lymphocytes from patients with COPD compared with those encountered in the control group. When whole blood from patients with COPD and their respective controls was perfused across CSE-stimulated HUAECs, increased and significant differences in leukocyte adhesion were observed, being more marked in the COPD group (figure 5E). Neutralisation of CX3CL1 activity on endothelial cells resulted in a significant reduction in CSE-induced leukocyte adhesion (52% inhibition in the control group and 84% inhibition in the COPD group, figure 5E). Despite these findings, no significant differences in the circulating levels of soluble CX3CL1 were encountered between the groups investigated (figure 5F).
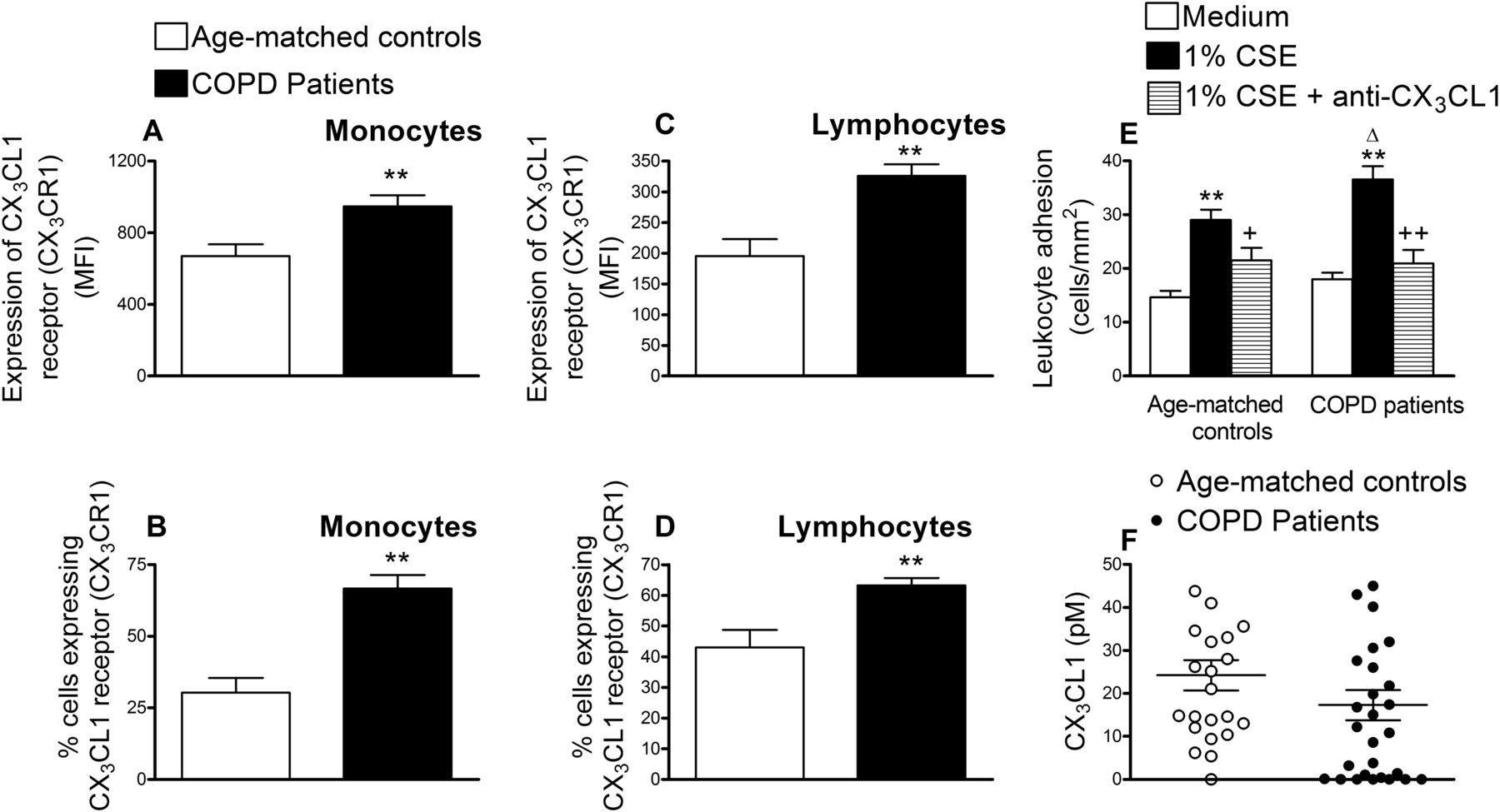
CX3CL1 receptor expression (CX3CR1) in circulating monocytes and lymphocytes from patients with chronic obstructive pulmonary disease (COPD) and aged-matched controls (A–D), recruitment of leukocytes from whole blood of patients with COPD and aged-matched controls by cigarette smoke extract (CSE)-stimulated human arterial umbilical endothelial cells (HUAECs) (E) and CX3CL1 plasma levels in patients with COPD and aged-matched controls (F). Leukocytes were stained with a conjugated monoclonal antibody for CX3CR1 and analysed by flow cytometry (A–D). Results are mean ± SEM of n = 23–29 experiments and are expressed as mean fluorescence intensity and percentage of positive cells. **p < 0.01 relative to values in the control group. HUAECs were stimulated with 1% CSE for 24 h. Some cells were incubated with a neutralising antibody against CX3CL1 function (5 µg/ml) or with an irrelevant isotype-matched monoclonal antibody (MOPC21, 5 µg/ml). Then, whole blood from patients with COPD and healthy aged-matched controls was perfused over the endothelial monolayers for 5 min at 0.5 dyn/cm2 and leukocyte adhesion quantified (E). Results are the mean ± SEM of n = 20–22 experiments. **p < 0.05 relative to values in the medium group. +p < 0.05 or ++p < 0.01 relative to 1% CSE group. Δ p < 0.05 relative to the values in the healthy aged-matched control group. CX3CL1 plasmatic levels were measured by ELISA (F). Results (pM in the plasma) are the mean ± SEM of n = 23–29 experiments.
CS exposure induces lung inflammation and leukocyte–endothelial cell interactions in the mouse cremasteric microcirculation; arteriolar leukocyte adhesion was reduced in CX3CR1−/− mice
Finally, to explore the potential in vivo relevance of these findings, an acute model of CS exposure was used. Histological examination of the lungs of animals exposed to CS for 3 days revealed a clear inflammatory response (figure 6A). Although significant cell recruitment was found in CS-exposed animals, no differences in lung leukocyte numbers were encountered between CX3CR1-expressing (CX3CR1−/+) and CX3CR1-deficient (CX3CR1−/−) mice (figure 6B). The recruited cells were mainly neutrophils (78.8±2.9%) and mononuclear cell numbers remained unchanged. In addition, no significant increases in CX3CL1 mRNA expression were detected (figure 6D).
Intravital microscopy was used to examine the effect of CS exposure on leukocyte–endothelial cell interactions in an organ remote from the lung, the mouse cremasteric microcirculation. CS exposure induced a significant enhancement of arteriolar leukocyte adhesion in CX3CR1−/+ and CX3CR1−/− mice (figure 6C) which was significantly reduced (51% inhibition) in CX3CR1-deficient mice (figure 6C). Mononuclear cells were found to be the cells primarily adhered to the CS-stimulated arterioles (86.6±3.4%). While mRNA quantification and immunofluorescence analysis of the cremasteric microcirculation revealed that endothelial CX3CL1 expression was virtually absent in the cremasteric arterioles of mice not exposed to CS, increased chemokine mRNA and protein expression was found in the microvessels of those animals exposed to the stimulus (figure 6E and F).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cigarette smoke (CS) exposure induces lung inflammation (A and B), leukocyte–arteriolar endothelial cell interactions (C) and increased CX3CL1 mRNA (D and E) and protein expression (E) in the mouse cremasteic microcirculation of CX3CR1-expressing (CX3CR1−/+) and CX3CR1-deficient (CX3CR1−/−) mice. Mice were exposed or left unexposed to CS for 3 days and responses examined 16 h later. Lungs were collected 16 h later and fixed for staining with hematoxylin/eosin (A) and cell counts were obtained (B). Leukocyte–arterial endothelium interactions were also determined (C). Results are mean ± SEM of n = 6–8 animals per group. *p < 0.05 or **p<0.01 relative to no exposed animals; +p<0.05 relative to CX3CR1−/+ mice. Columns show the fold increase in lung (D) and cremaster (E) CX3CL1 mRNA expression relative to control β actin. Results are mean ± SEM of n = 4 independent experiments. **p < 0.01 relative to no exposed animals. Some cremaster muscles were fixed for CX3CL1 and endothelium (CD31) staining (F). CX3CL1 expression is shown in green (stained with an Alexa Fluor 488-conjugated donkey anti-rabbit secondary antibody) and vessel endothelium (red) was stained with a PE-conjugated anti-mouse CD31 monoclonal antibody. Overlapping expression of CX3CL1 and CD31 is shown in yellow. Results are representative of n = 5–6 animals per group.
Discussion
Cardiovascular diseases are more frequent and are found prematurely in patients with chronic inflammatory disorders such as COPD.1 However, little is known regarding the mechanisms by which CS induces endothelial dysfunction in organs distant from the lung. In this study, we demonstrated for the first time that CSE induces CX3CL1-dependent mononuclear cell arrest in the arterial endothelium and unravels a previously undescribed mechanism by which CS affects arteriolar function in an adverse manner. Different human lymphocyte subsets as well as monocytes express CX3CR1 receptor,21 which might explain the marked reduction in mononuclear cell adhesion to CSE-stimulated HUAECs after neutralising CX3CL1 activity.
Smoke-derived free radicals and oxidants are part of CS causing a pro-oxidative state in the circulatory system. CS exposure rapidly induces production of ROS impairing endothelial functions.22 Moreover, CX3CL1 expression can be upregulated by oxidative stress23 and we now report the involvement of ROS in CS-induced CX3CL1 expression and mononuclear leukocyte arrest since apocynin, an unspecific Nox inhibitor, diminished these responses. Based on these results, we tried to clarify the endothelial Nox isoforms involved in these findings. Endothelial cells mainly express Nox2, Nox4 and Nox5 isoforms17 ,18 and while CSE stimulation of HUAECs caused increased Nox2 and Nox4 expression, their knockdown did not significantly impact CSE-induced responses. Conversely, Nox5 downregulation was able to abrogate CSE-induced CX3CL1 expression and subsequent mononuclear cell adhesion. Nevertheless, it cannot be excluded that the potential interaction of Nox5 with other Nox isoforms in the context of Nox5 downregulation, which may inhibit Nox5-associated Nox activity, although such contention requires further studies. Of note, a recent report has demonstrated the involvement of Nox5 in angiotensin-II-induced increased endothelial CAM expression,24 a mediator that shares a similar profile of inflammatory effects with CS since both ROS generation and TNFα release seem to be involved in its proinflammatory activity.25 ,26
The role of TNFα in COPD is thought to be central to lung and systemic inflammation.1 The potential implication of TNFα was investigated and CSE stimulation clearly increased the expression of this cytokine. We also revealed that TNFα silencing in HUAECs was associated with reductions in CSE-induced CX3CL1 expression with concomitant impairment in CSE-induced mononuclear leukocyte–endothelial cell interactions. Taken together, these findings suggest that CS contributes to endothelial dysfunction and vascular damage through TNFα release, which may exert autocrine/paracrine effects in the arterial endothelium and probably in the vascular smooth muscle cells via increased CX3CL1 expression and subsequent leukocyte CX3CR1–endothelial CX3CL1 interactions.
Activation of MAPK signal transduction is important to stress-induced gene expression; such stresses include CS and TNFα.20 We found that CSE-mediated activation of endothelial cells triggers different redox-sensitive signalling pathways likely activated by NADPH oxidases such as p38 MAPK and NFκB. Furthermore, previous studies have reported a requirement of p38 MAPK and NFκB activation in the increased CX3CL1 expression23 ,27 and here we show through the blockade of p38 MAPK and NFκB signalling by pharmacological inhibition that CSE-induced activation of these signalling pathways is indispensable for endothelial CX3CL1 expression and its leukocyte-capturing function. Based on the evidence that the transcriptional activity of NFκB is regulated among others by p38 MAPK pathways,28 it is likely that this kinase acts upstream of NFκB activation. Additionally, the human CX3CL1 promoter contains a number of putative DNA binding elements, including ones for NFκB.29
To determine if these results have any clinical relevance we examined mononuclear cells from patients with COPD. Our data demonstrated CX3CR1 overexpression and increased circulating numbers of CX3CR1+ monocytes and lymphocytes in patients with COPD compared with those from their age-matched controls. Consequently, leukocyte adhesion to CSE-stimulated HUAECs was more pronounced in the COPD group. Moreover, although CX3CL1 neutralisation reduced the adhesion of CX3CR1+ cells to the arterial endothelium in both groups, this effect was more marked when whole blood was from patients with COPD (52% vs 84% inhibition). Several explanations may account for the clinical impact of these results. First, an identified mutant form of the CX3CL1 receptor, termed CX3CR1-M280, is defective in mediating adhesive and chemotactic activity.30 ,31 This mutated form of CX3CR1 is linked to lower risk of atherosclerosis, acute coronary events and coronary artery endothelial cell dysfunction.30 ,31 Second, CX3CR1 upregulation was detected in circulating monocytes of patients with coronary artery disease.32 Therefore, it is feasible that increased CX3CR1 expression/function in circulating mononuclear cells may establish a direct link between COPD and the development of cardiovascular disorders.
These striking observations prompted us to evaluate in vivo the impact of CS exposure. In our study CS exposure resulted in a moderated lung inflammation characterised by leukocyte infiltration in the lung tissue. However, no differences between both strains of animals (CX3CR1 expressing vs CX3CR1 deficient) were detected. This is not surprising given that neutrophils were the primary leukocytes involved in these responses and they do not express CX3CR1 receptor.21 Likely a chronic exposure to this stimulus is required for the accumulation of CX3CR1+ cells within the lung parenchyma as found in the past33 since in our acute model no significant increase in lung CX3CL1 mRNA expression was detected. Previous reports have indicated that even moderate cigarette smoking leads to circulating monocyte activation and their increased adhesion to the endothelium.34 Here we showed that CS-induced increased CX3CL1 expression is relevant for the attachment of mononuclear cells to the arterial endothelium. Unlike human monocytes, murine monocytes are the main subtype of leukocytes expressing CX3CR1.35 Monocytes with high CX3CR1 expression were found to patrol blood vessels and extravasate rapidly in response to damage or infection as part of the early inflammatory response.35 Furthermore, monocytes are abundant in atherosclerotic lesions and CX3CR1 has been implicated in the pathogenesis of this inflammatory disease.11 ,12 Thus, it seems that acute exposure to CS causes endothelial dysfunction in the lung manifested as neutrophil infiltration and leukocyte–endothelial cell interactions in organs distant from the lung, resulting in CX3CL1-dependent arteriolar monocyte adhesion.
In conclusion, we have provided evidence that CS induces CX3CL1-dependent mononuclear cell arrest by arterial endothelium in vitro and in vivo. To our knowledge, this is the first report showing a previously undescribed mechanism that may account for the increased development of cardiovascular disorders in smokers. Our study also provides new insights into potential cellular and molecular mechanisms underlying these responses. Furthermore, we have proved the potential clinical implications of our findings since blockade of the CX3CL1/CX3CR1 axis dramatically reduced the adherence of mononuclear leukocytes from patients with COPD to CSE-stimulated arterial endothelium. Therefore, CX3CL1 and CX3CR1, and chemokine-regulatory factors such as TNFα may be considered as potential drug targets for the prevention and treatment of COPD-associated cardiovascular disease.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CR, CC, LP, MCN, CG, ES, EM and MJS participated in the acquisition of the data, and the analysis and interpretation of the results. AL contributed with vital reagents and was involved in the design of the study and in its revision prior to submission. EM was also involved in the design of the study and in its revision prior to submission. MJS was involved in the conception, hypothesis delineation and design of the study as well as in the article writing. CR and CC contributed equally.
-
Funding This study was supported by grants SAF2008-03477, SAF2009-08913, SAF2011-23777, CP07/00179 and PI081875, Spanish Ministry of Science and Innovation, RIER RD08/0075/0016, Carlos III Health Institute, Spanish Ministry of Health, the European Regional Development Fund (FEDER) and research grants from Generalitat Valenciana (PROMETEO/2008/045; AP-055/08; GVACOMP2010-129). CC and CR were supported by a grant from the Spanish Ministry of Science and Innovation.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Institutional ethics committee at the University Clinic Hospital of Valencia, Spain.
-
Provenance and peer review Not commissioned; externally peer reviewed.