Article Text

Abstract
Background The purpose of this randomised double-blind double-dummy placebo-controlled trial was to investigate whether etanercept, a tumour necrosis factor α (TNFα) antagonist, would provide more effective anti-inflammatory treatment for acute exacerbations of chronic obstructive pulmonary disease (COPD) than prednisone.
Methods We enrolled 81 patients with acute exacerbations of COPD and randomly assigned them to treatment with either 40 mg oral prednisone given daily for 10 days or to 50 mg etanercept given subcutaneously at randomisation and 1 week later. Both groups received levofloxacin for 10 days plus inhaled bronchodilators. The primary endpoint was the change in the patient's forced expiratory volume in 1 s (FEV1) 14 days after randomisation. Secondary endpoints included 90-day treatment failure rates and dyspnoea and quality of life.
Results At 14 days the mean±SE change in FEV1 from baseline was 20.1±5.0% and 15.2±5.7% for the prednisone and etanercept groups, respectively. The mean between-treatment difference was 4.9% (95% CI −10.3% to 20.2%), p=0.52. Rates of treatment failure at 90 days were similar in the prednisone and etanercept groups (32% vs 40%, p=0.44), as were measures of dyspnoea and quality of life. Subgroup analysis revealed that patients with serum eosinophils >2% at exacerbation tended to experience fewer treatment failures if treated with prednisone compared with etanercept (22% vs 50%, p=0.08).
Conclusions Etanercept was not more effective than prednisone for treatment of acute exacerbations of COPD. Efficacy of prednisone was most apparent in patients who presented with serum eosinophils >2%.
Clinical Trials gov number NCT 00789997.
- COPD Exacerbations
- COPD Pharmacology
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does treatment of acute exacerbations of COPD with the TNFα antagonist etanercept provide safer, more effective, anti-inflammatory treatment than prednisone?
What is the bottom line?
-
Etanercept was not found to be more effective than prednisone for treatment of acute exacerbations of COPD.
Why read on?
-
Subgroup analyses suggest that patients with peripheral blood eosinophilia at time of AECOPD were those who primarily benefited from oral prednisone, whereas those without eosinophilia did equally well with etanercept or steroids.
Introduction
Acute exacerbations of chronic obstructive pulmonary disease (AECOPD) are characterised clinically by symptoms of worsening dyspnoea, cough, sputum production and sputum purulence, as well as by worsening of airflow obstruction.1 The onset of an exacerbation is often sudden, and median symptom recovery times average 11–13 days.2 Standard treatment for AECOPD includes treatment with antibiotics and corticosteroids.3 Clinical trials have demonstrated that the addition of corticosteroids to antibiotic therapy decreases treatment failure rates in inpatients with AECOPD4 and prevents relapse in outpatients with AECOPD.5 However, 27% of patients fail treatment with combined antibiotic and corticosteroid therapy by 30 days, and treatment failure rates rise to 37% by 90 days.4 ,5
A recent clinical trial by Bafadhel et al6 suggested that the peripheral blood eosinophil count might be a useful biomarker to direct corticosteroid therapy during AECOPD. The trial suggested that some patients with AECOPD may not benefit from treatment with oral corticosteroids. In their trial, patients with blood eosinophils ≤2% exhibited greater improvements in quality of life and fewer treatment failures if they were treated with placebo rather than with prednisolone.
Exacerbations of COPD are inflammatory events, and during exacerbations there are marked increases in airway neutrophils7–9 with increases in sputum concentrations of tumour necrosis factor α (TNFα), CXCL8 and interleukin 6 (IL-6).8 ,10 ,11 TNFα appears to play a major role in driving inflammation in AECOPD, and concentrations in sputum rise more than fourfold during exacerbations compared with the stable state.10 TNFα upregulates adhesion molecules and facilitates migration of leucocytes into the bronchial mucosa during AECOPD by inducing IL-8 expression, and it stimulates neutrophil degranulation and superoxide production.12
Etanercept exerts its pharmacological effect by binding specifically to soluble TNF and by blocking the interaction of TNF with cell surface TNF receptors. The drug competitively inhibits TNF binding to cell surface TNF II receptors, rendering TNF biologically inactive.13 A recent observational study found that use of etanercept in a cohort of patients who were diagnosed with both rheumatoid arthritis and COPD was associated with a decreased risk of hospitalisation for AECOPD (RR 0.49; 95% CI 0.29 to 0.82).14 The authors suggested their results supported the initiation of randomised trials of etanercept in patients with COPD at high risk for exacerbations.15
Standard treatment with high-dose corticosteroids for AECOPD has a high failure rate and is often associated with adverse effects such as osteoporosis, glucose intolerance and psychiatric side effects.14 ,16 Given the above limitations of standard treatment, an alternative anti-inflammatory treatment for AECOPD that is more efficacious and potentially less toxic would be desirable. We therefore conducted a randomised controlled clinical trial comparing etanercept against prednisone for treatment of AECOPD. Our primary objective was to determine if etanercept improved lung function and decreased treatment failure rates to a greater extent than prednisone.
Methods
Study design
This was a randomised double-blind double-dummy multicentre controlled clinical trial incorporating two parallel treatment arms. Patients underwent study assessments on the day of the exacerbation and at 7, 14, 30 and 90 days after randomisation. The research ethics boards of all the participating hospitals approved the study and all patients gave signed informed consent prior to study entry.
Patients
We enrolled patients with AECOPD who presented urgently to a physician or to the emergency department at eight Canadian academic medical centres. Enrolled patients fulfilled at least two of the following three clinical criteria for COPD exacerbation: increased dyspnoea, sputum volume, sputum purulence.1
Only those patients with a previous diagnosis of COPD established by a physician and airflow obstruction on spirometry, defined as forced expiratory volume in 1 s (FEV1) ≤ 70% predicted and an FEV1/forced vital capacity (FVC) ratio ≤70% were eligible. Additional inclusion criteria necessary for enrolment were age >35 years and a history of cigarette smoking of ≥10 pack-years.
We excluded patients who had received more than a single dose of oral or injectable corticosteroids in the 30-day period preceding trial entry. Patients with respiratory failure necessitating use of invasive or non-invasive mechanical ventilation and patients with a history of asthma or other chronic lung diseases were excluded. A chest x-ray was taken prior to randomisation and patients with pneumonia or congestive heart failure or suspected malignancy on the x-ray were excluded. Patients with a history of tuberculosis, non-tuberculous mycobacteria or fungal infection were excluded, as were patients with a history of multiple sclerosis or demyelinating disease, HIV, hepatitis B or hepatitis C, or those with malignancy within the past 5 years. We also excluded pregnant or nursing mothers, patients with serum white blood cell (WBC) count ≤3000 or serum WBC ≥ 20 000 and patients with a temperature ≥38.5° at presentation.
Study intervention
Patients were randomly allocated to either prednisone 40 mg taken orally once daily for 10 days or to subcutaneous etanercept 50 mg given by the study research nurse or physician on the day of randomisation and 7 days later. Patients randomised to prednisone received placebo subcutaneous injections of sterile saline at randomisation and on day 7, which were identically labelled and identical in appearance to the etanercept injections. Patients randomised to the etanercept arm received placebo prednisone capsules for 10 days which were identical in appearance to the prednisone capsules used in the trial. Patients in both treatment groups were treated with open-label levofloxacin 750 mg daily for 10 days, or an alternative antibiotic if the patient was allergic or intolerant to levofloxacin. Patients in both groups were also prescribed an inhaled long-acting β-agonist (either salmeterol or formoterol) and an inhaled long-acting anticholinergic bronchodilator (tiotropium) after randomisation. If the patient was using an inhaled steroid or inhaled steroid/long-acting β-agonist combination product at the time of randomisation, this medication was continued for patients in both groups. Supplemental salbutamol was provided as needed throughout the trial period.
We chose to use the dose of etanercept recommended for adults with rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis (50 mg given once weekly). Pharmacokinetic studies suggest that the absolute bioavailability of etanercept is 58% following a single 50 mg subcutaneous dose, and that etanercept peak concentrations of 1460 ng/ml are achieved within 2–3 days after a single 50 mg dose.17 Other studies have shown that, at peak serum concentrations following a 50 mg dose, free TNFα is completely bound by etanercept and is therefore unable to interact with cell surface receptors.13 Etanercept has a half-life of approximately 70 h in normal patients and 100 h in those with rheumatoid arthritis. Therefore, because of its relatively slow elimination, two subcutaneous injections given 1 week apart on day 0 and day 7 of the study would provide treatment with appropriate therapeutic blood levels necessary for neutralisation of TNFα over a 10–14-day period.17
Randomisation was by central allocation of a randomisation schedule prepared through a computer-generated random listing of the two treatment allocations blocked by variable blocks of two or four and stratified by site. Study medications were dispensed by the site research pharmacist according to the patient's randomisation assignment. Neither the research staff, patients, nor the treating physicians were aware of the treatment assignment before or after randomisation. Patients were taught correct bronchodilator inhalation technique on the day of randomisation.
Outcome measures
The primary outcome was the change in FEV1 from baseline, measured from the day of randomisation to 14 days after randomisation. FEV1 was obtained using calibrated spirometers at approximately the same time of day at all visits throughout the study. It was assessed on the day of randomisation after the patient had received emergency treatment for their acute exacerbation in the emergency department or outpatient clinic. On subsequent study visits, FEV1 was assessed after the patient has taken the morning prescribed dose of anticholinergic and long-acting β-agonist medications. Up to five FEV1 measurements were obtained in an effort to achieve three acceptable efforts. The highest acceptable FEV1 and the highest FVC measurement each obtained on any of three blows (even if not from the same curve) meeting the American Thoracic Society criteria constituted the data for that test set.
Secondary outcomes included the proportion of treatment failures occurring within 90 days after the onset of exacerbation and the time to treatment failure. Treatment failure was defined according to Niewoehner et al4 as the need for intensification of therapy with open-label systemic glucocorticoids, or the need for hospitalisation (for outpatients) or rehospitalisation (for inpatients), or the need for mechanical ventilation or death. For each suspected treatment failure we contacted both the patient and the patient's treating physician and obtained a copy of the written medical record of the medical encounter (when available) to ensure that the event met the study definition of treatment failure.
Other secondary outcomes included changes in FEV1 at days 7, 30 and 90 after randomisation and changes in health-related quality of life and dyspnoea. Health-related quality of life was assessed with the Chronic Respiratory Questionnaire (CRQ),18 which has been validated for use in patients with AECOPD.19 Dyspnoea was assessed with the Baseline and Transitional Dyspnoea Index20 and with the dyspnoea domain of the Chronic Respiratory Disease Questionnaire.21 Changes in serum levels of C-reactive protein and changes in circulating serum TNF and TNF receptor I (55 kDa) and TNF receptor II (75 kDa) levels were assessed using high-sensitivity ELISA with signal amplification (eBioscience, Vienna, Austria and R&D Systems, Minneapolis, Minnesota, USA).
Complete blood counts, renal function, liver function and blood glucose were assessed at 7 and 14 days after randomisation for safety monitoring. Serious adverse events—including incidence of pneumonia, opportunistic infections, malignancy and death—were assessed at each follow-up study visit up to day 90.
Statistical analysis
We designed this study to detect an absolute between-group difference in improvement in FEV1 of 0.20 l from baseline to 14 days. We assumed a normal distribution for improvement in FEV1, a two-sided α error of 0.05, a β error of 0.20, a SD of 0.30 l in improvement for the two groups and a conservative assumption of no correlation between baseline FEV1 and change in FEV1. Under these assumptions, 74 subjects were required. The total sample size was increased to 80 subjects to allow for an 8% rate of loss to follow-up.
The final analysis was conducted on an intention-to-treat basis. All significance tests were two-tailed. The principal analysis of the relative change in FEV1 from baseline to 14 days was conducted using an unadjusted t test. Patients were included in the analysis according to the group to which they were randomised, regardless of crossover or compliance. The proportion of patients who were treatment failures at 90 days was analysed using the χ2 test and the time to treatment failure was analysed using Kaplan–Meier survival curves and a log-rank test. Transitional Dyspnoea Index scores and changes in the scores of the components of the CRQ were analysed using parametric t tests.
An interim analysis of efficacy was not performed. An independent Data Safety Monitoring Board did oversee the trial and monitor all adverse events on a quarterly basis but did not advise early stopping at any point during the study.
Results
Study population
A total of 166 potential patients were screened for the study (figure 1) and 81 patients were randomised; 41 received etanercept and 40 received prednisone. The two treatment groups were similar with respect to baseline characteristics, although the prednisone-treated group included six inpatients whereas only one inpatient was randomised to the etanercept group (table 1). Four patients withdrew prematurely from the study and one died before reaching the primary endpoint which was measured on day 14 after randomisation (figure 1). Two patients discontinued study treatment early but stayed in the study. Ninety-day treatment failure data were available and assessed in 78 of the 81 randomised patients (96%).
Baseline characteristics of the 81 randomised patients according to treatment assignment
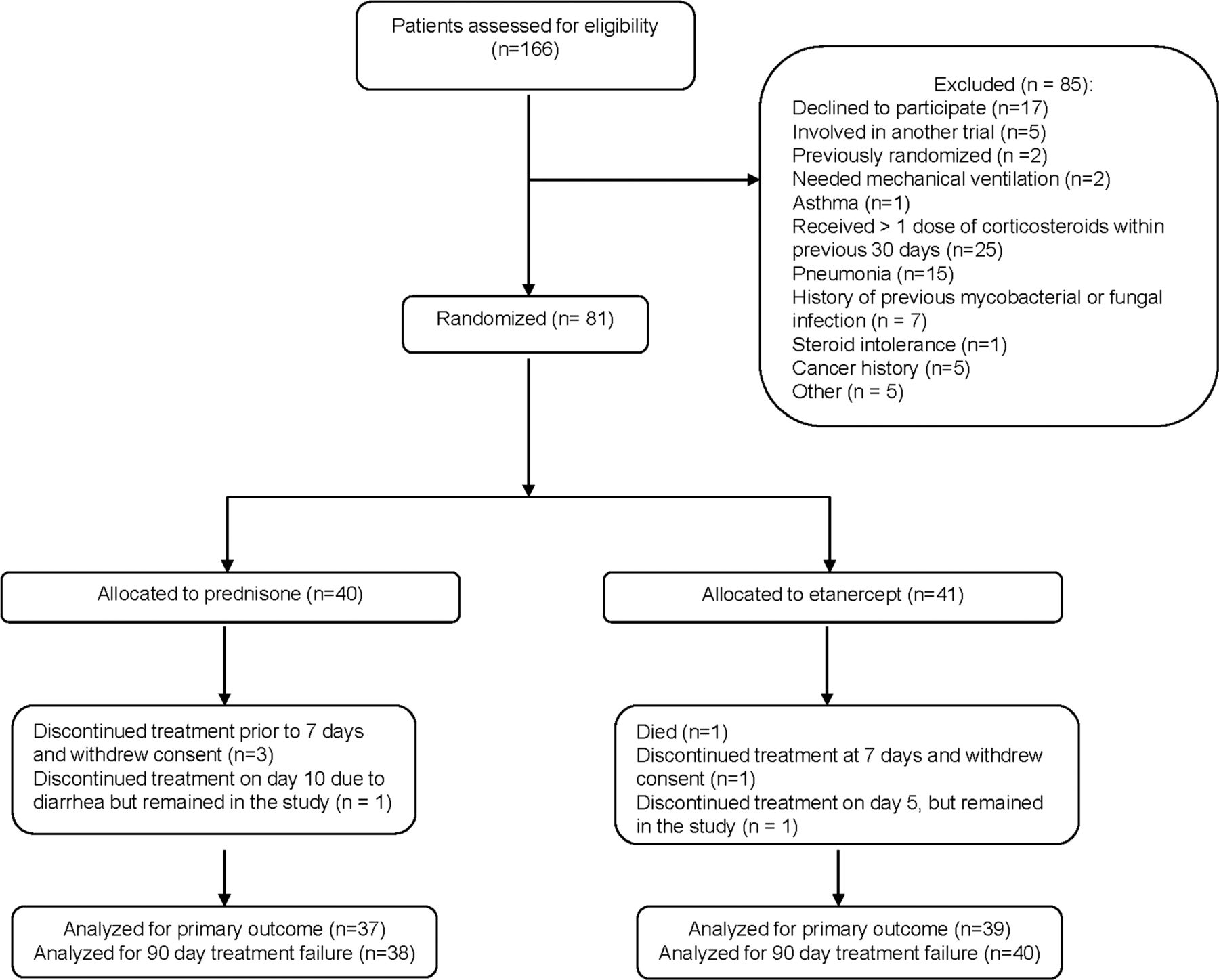
Trial profile.
Primary outcome
The absolute change in FEV1 from baseline to 14 days was 0.139 l (95% CI 0.004 to 0.275) in the etanercept-treated patients and 0.164 l (95% CI 0.086 to 0.241) in the prednisone-treated group. The mean between-group treatment difference was 0.024 l (95% CI −0.130 to 0.179), p=0.75. After 14 days the mean±SE change in FEV1 from baseline was 15.2±5.7% and 20.1±5.0% for the etanercept and prednisone groups, respectively. The mean between-group treatment difference was 4.9% (95% CI −10.3% to 20.2%) in favour of the prednisone group (p=0.52, figure 2).

Changes in forced expiratory volume in 1 s (FEV1) from baseline in the two treatment groups. Black solid line represents etanercept group, blue dashed line represents prednisone group.
Secondary outcomes
Changes from baseline in FEV1 were not statistically different between the two treatment groups at 7, 30 or 90 days. Between-group treatment differences (prednisone minus etanercept) were −1.3% (95% CI −13.0% to 10.4%), 0.7% (95% CI −12.7% to 14.1%) and 2.6% (95% CI −10.4% to 15.6%), respectively (figure 2).
The proportion of patients who experienced a treatment failure by 90 days was not significantly different between the etanercept and prednisone groups (table 2). In the etanercept group 16/40 (40%) failed treatment compared with 12/38 (32%) in the prednisone group (p=0.44). Time to treatment failure was not significantly different between the two groups (log-rank p=0.38, figure 3).
Ninety-day treatment failure outcome according to treatment assignment
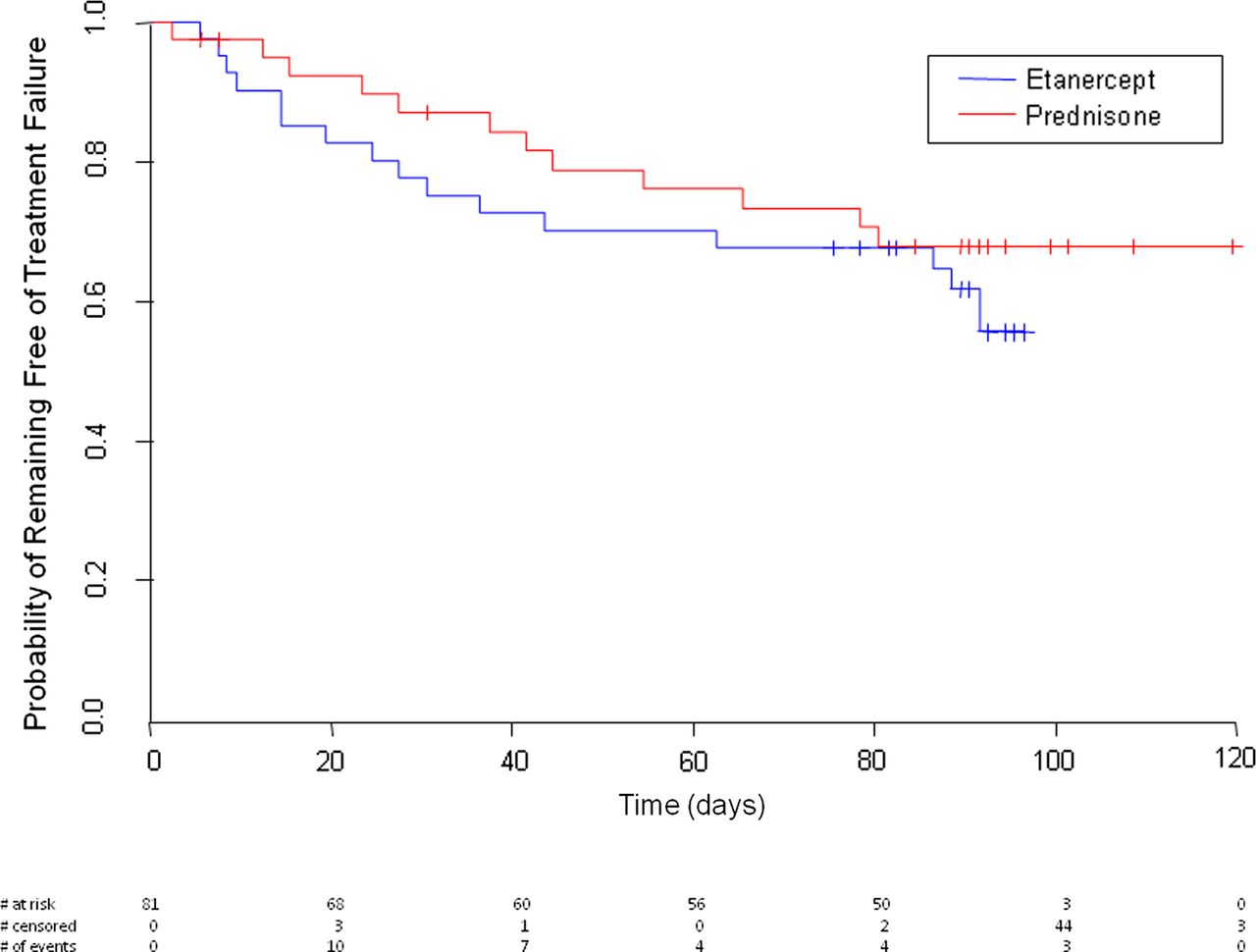
Kaplan–Meier estimates of the probability of remaining free of treatment failure according to treatment assignment. Time to treatment failure was not significantly different between the two groups (log-rank p=0.38). #, censored observations.
Patients in both treatment groups experienced improvements in dyspnoea at 14 days and 90 days after randomisation as measured by positive scores in the Transitional Dyspnoea Index and by the dyspnoea domain of the CRQ. Improvements were not significantly different between the groups (table 3).
Changes in dyspnoea and quality of life according to treatment assignment
Changes in disease-specific quality of life were not significantly different at 14 days and 90 days after randomisation in the two treatment groups for all four domains of the CRQ. Higher scores indicate greater improvements in quality of life as measured by the questionnaire (table 3). The only exception was that patients treated with prednisone showed a greater improvement in scores on the mastery domain at 14 days than those treated with etanercept with a between-treatment difference of 0.77 points (95% CI 0.16 to 1.38, p=0.015).
Inflammatory markers
C-reactive protein declined in both groups after 14 days; however, the changes were highly variable and there was no significant difference between the groups (table 4). Serum TNFα and TNF receptor II levels increased significantly after 14 days in the etanercept-treated patients compared with those treated with prednisone (table 4).
Changes in inflammatory markers from baseline to 14 days according to treatment assignment
Safety
Eleven patients experienced serious adverse events during the 90-day study period, four in the etanercept group and seven in the prednisone group (table 5). There was one study-associated death in a patient in the etanercept group who suffered a sudden cardiopulmonary arrest 3 days after study entry. One patient in the prednisone group was diagnosed with lung cancer during the 90-day study period. No cancers were recorded in the patients in the etanercept group. There were two cases of pneumonia in the etanercept group and three in the prednisone group. Six patients developed Achilles tendonitis, four in the prednisone group and two in the etanercept group. Overall, etanercept was well tolerated and there were no unexpected safety findings in this study population.
Adverse events
Subgroup analysis based on blood eosinophils
Eighteen of 41 patients (44%) in the etanercept group and 18 of 40 patients (45%) in the prednisone group had serum eosinophil counts which were >2% of the total circulating WBC count. The mean between-group treatment difference in FEV1 after 14 days in this subgroup was 12.2% (95% CI −17.7% to 42.1%) in favour of the prednisone-treated patients. In the subgroup of patients with eosinophils >2%, only four (22%) treated with prednisone failed treatment compared with nine (50%) treated with etanercept (p=0.08, figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Subgroup analysis: treatment failures grouped by treatment assignment and blood eosinophil count.
In contrast, in the subgroup with eosinophils ≤2%, responses to etanercept and prednisone were more similar between the two treatment groups. The mean between-group treatment difference in FEV1 after 14 days in this subgroup was 2.6% (95% CI −9.9% to 15.1%) in favour of the etanercept-treated patients. In the subgroup with eosinophils ≤2%, treatment failure rates were 42% in those treated with prednisone and 33% in those treated with etanercept (p=0.57, figure 4).
An alternative subgroup analysis based on an absolute eosinophil count of >200 eosinophils ×109/l yielded the same trend to better outcomes in those with greater blood eosinophilia who received prednisone. In the subgroup with eosinophil count >200×109/l, 3/10 patients (30%) treated with prednisone failed treatment compared with 7/13 (54%) treated with etanercept (p=0.26). The relatively small size of these subgroups limits definite statistical conclusions.
Discussion
This is the first randomised controlled trial to evaluate the use of TNFα antagonists for the treatment of AECOPD. Our study evaluated the efficacy and safety of etanercept 50 mg injected once weekly for 2 weeks against standard treatment with prednisone 40 mg daily for 10 days. Etanercept failed to demonstrate evidence of improved clinical efficacy compared with standard treatment with prednisone for any of the lung function or clinical endpoints evaluated during this trial. Improvements, while not statistically different between the two treatment groups, tended to favour the prednisone-treated patients.
To date, only one large trial of TNF antagonists for the treatment of chronic stable COPD has been published.22 This study, by Rennard and colleagues, was a multicentre controlled trial that randomised stable patients with moderate to severe COPD to treatment with the anti-TNF antibody infliximab or to placebo. After 44 weeks of treatment the infliximab-treated patients did not demonstrate improvements in CRQ scores or in FEV1, 6 min walk distance or dyspnoea. The trial investigators concluded that TNFα antagonists were not beneficial in patients with stable COPD.22
Our study differs from that by Rennard et al in that we studied patients with AECOPD rather than stable COPD. Patients were placed on short-term treatment with etanercept and the responses to treatment were compared against the current standard of care therapy with prednisone rather than placebo. Although observational studies and laboratory-based studies had suggested that etanercept could protect against COPD exacerbations, our randomised controlled trial was unable to show a benefit of etanercept compared with prednisone for the treatment of acute exacerbations.
Administration of etanercept results in significant increases in serum levels of circulating TNFα and TNF receptor II.23 As expected, these increases were seen in patients who received etanercept in the current study. However, the increased circulating TNF has been shown to be biologically inactive since it is bound to the soluble etanercept TNF receptor agonist and is unable to bind to TNF receptors on cellular membranes.23
Our study does have several limitations. In light of the recently published study from Bafadhel et al,6 ,24 it would have been advantageous if we had included only patients with non-eosinophilic COPD exacerbations in our trial since this subgroup of patients may be expected to have a poorer response to prednisone and perhaps a favourable response to alternative non-steroid anti-inflammatory agents directed against neutrophilic inflammation. Unfortunately, the trial by Bafadhel was published in 2012, after our trial had already completed recruitment, and our trial did not limit recruitment to a non-eosinophilic subgroup.
This study was designed as a proof-of-concept study meant to support a potential second larger definitive study. As such, our sample size was not large, and our study was powered to show a difference in change in lung function over 14 days rather than 90-day treatment failure rates, which would have been a more important clinical outcome. A further limitation of our study is that the improvement in FEV1 in both groups was lower than had been expected from previous trials and was therefore lower than the between-group difference on which we based the study sample size. The lower than expected improvements in FEV1 seen in both groups may have limited our ability to observe realistic FEV1 differences between the two groups.
Our intent was to determine, as a first step, whether etanercept therapy improved lung function to a greater extent than prednisone and to determine whether etanercept was safe to use in the setting of AECOPD. If efficacy and safety had been demonstrated relative to standard therapy with prednisone, then we would have proceeded to a second large definitive trial to determine whether etanercept could decrease 90-day treatment failure rates. However, although we did not observe any safety concerns during our study, the results of our trial did not suggest that etanercept was more effective at improving FEV1 or other clinical outcomes compared with prednisone, and the relative risk of 90-day treatment failure was greater (by 25%) in those randomised to etanercept. Based on the results of this study, a second large clinical trial of etanercept in unselected patients with COPD exacerbation is probably not warranted although further trials in patients who do not have serum eosinophilia could be of benefit.
Acknowledgments
The authors would like to thank the following people for their invaluable assistance: Study coordinators: Gay Pratt, Isabelle Depault, Ottawa Hospital Research Institute, Ottawa; Marie-Josee Breton, Marthe Bélanger, Brigitte Jean, Josée Picard, Centre de recherche de l'Institut universitaire de cardiologie et de pneumologie de Québec, Québec; Jimmy Joy, Carolyn Robertson, Curtis Dumonceaux, University of Calgary, Calgary; Rommel Mangaser, Leo Cicora, McGill University Health Centre, Montreal; Janet Baron, University of Saskatchewan, Saskatoon; Tyrone Maguire, St Paul's Hospital, Vancouver; Linda Hui, Giselle Gerdak, University of British Columbia, Vancouver; Melanie Kjarsgaard, St Joseph's Healthcare Hamilton, Hamilton. Pharmacy: Anne-Marie Dugal, Susan Fetzer, The Ottawa Hospital, Ottawa; Nathalie Châteauvert, Centre de recherche de l'Institut universitaire de cardiologie et de pneumologie de Québec, Québec; Christine Wallace, St Joseph's Healthcare Hamilton, Hamilton; and the staff from St Paul's Hospital, Vancouver; McGill University Health Centre Montreal, Québec; and the University of Saskatchewan, Saskatoon. Data managers: My-Linh Tran, Jennie Cote, Ottawa Hospital Research Institute, Ottawa. Data Safety Monitoring Board: Dr Maureen Meade, Dr Matthew Stanbrooke, Dr Charlene Fell.
References
Footnotes
-
Contributors SDA, KLV, FM, JB and JMFG contributed to the conception and design of this study. SDA, KLV, FM, JB, SKF, DDS, DDM, JMFG and PN contributed to the acquisition of data. SDA and RM contributed to the analysis and interpretation of data. SDA drafted the manuscript and all authors critically revised it for important intellectual content and approved the final version to be submitted.
-
Funding Funded by Canadian Institutes of Health Research grant MCT-901676.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement SDA has all of the raw data related to this study in his possession and is willing to share all data with the study editors or with other investigators.