Article Text

Abstract
Airway epithelial cells and immune cells participate in the inflammatory process responsible for much of the pathology found in the lung of patients with cystic fibrosis (CF). Intense bronchial neutrophilic inflammation and release of proteases and oxygen radicals perpetuate the vicious cycle and progressively damage the airways. In vitro studies suggest that CF transmembrane conductance regulator (CFTR)-deficient airway epithelial cells display signalling abnormalities and aberrant intracellular processes which lead to transcription of inflammatory mediators. Several transcription factors, especially nuclear factor-κB, are activated. In addition, the accumulation of abnormally processed CFTR in the endoplasmic reticulum results in unfolded protein responses that trigger ‘cell stress’ and apoptosis leading to dysregulation of the epithelial cells and innate immune function in the lung, resulting in exaggerated and ineffective airway inflammation. Measuring airway inflammation is crucial for initiating treatment and monitoring its effect. No inflammatory biomarker predictive for the clinical course of CF lung disease is currently known, although neutrophil elastase seems to correlate with lung function decline. CF animal models mimicking human lung disease may provide an important insight into the pathogenesis of lung inflammation in CF and identify new therapeutic targets.
- Bacterial Infection
- Cystic Fibrosis
- Cytokine Biology
- Bronchiectasis
- Bronchoscopy
- Exhaled Airway Markers
Statistics from Altmetric.com
Introduction
The pathophysiology of cystic fibrosis (CF) lung disease is the consequence of a cascade of events initiated by CF transmembrane conductance regulator (CFTR) dysfunction that causes failure of chloride secretion and sodium hyperabsorption at the apical airway surface, leading to dehydration of the airway surface fluid layer and impaired mucociliary clearance. The desiccated secretions obstruct the airways and prevent elimination of bacteria from the lung, permitting bacterial infection to become established. The resultant neutrophilic inflammation causes progressive damage to the conducting airways. The inflammatory response in the lungs of patients with CF is self-perpetuating; neutrophil-derived mediators fuel the process and induce further recruitment of inflammatory cells.1 The vicious cycle of intense neutrophilic inflammation, oxidative stress and overwhelmed defences amplifies inflammation, perpetuates infection and contributes to irreversible airway destruction and fibrosis. Cellular apoptosis and airway remodelling ensue with irreversible lung damage. Despite advances in our understanding of the molecular and cellular basis of CF, there remains a paradox of why recruited neutrophils fail to eradicate bacterial infections in the lung. Moreover, why do CF airways contain high levels of proinflammatory mediators that continuously recruit neutrophils into the endobronchial lumen, even in the absence of clinically apparent infection?2S1
There is accumulating evidence, mainly from in vitro studies, suggesting that CFTR dysfunction may directly affect airway immunity through increased production of proinflammatory mediators and impaired immune responses to pathogens. These studies suggest that CFTR-related dysregulation of respiratory epithelial cells and innate immune dysfunction in the lung leads to an exaggerated and ineffective airway inflammation which fails to eradicate pulmonary pathogens.1 This review provides a summary of newer insights into the development of the inflammatory process in CF and its relationship with the irreversible and progressive damage to the lungs of patients with CF.
Development of inflammation in CF airways
In patients with CF the airways are considered normal at birth with the exception of plugging and distension of submucosal gland ducts, suggesting that some changes to the airways may occur prenatally.S2 Emerging evidence suggests that lung disease begins very early in life. Infants diagnosed by newborn screening demonstrated the presence of inflammation and bacterial infection, especially with Staphylococcus aureus, soon after diagnosis.3 S3 Inflammatory markers such as interleukin 8 (IL-8), peroxidases and their oxidants were found in high concentrations in bronchoalveolar lavage (BAL) fluid, even in infants with normal lung function and without apparent bacterial colonisation.3 S3 In another study IL-8 was elevated in most infants with CF diagnosed by newborn screening, the presence of respiratory symptoms at the time of BAL was associated with elevated neutrophil count and more free neutrophil elastase (NE) activity but not with infection.S4 In contrast, other studies suggest that infection precedes inflammation in infants with CF and that uninfected control subjects have similar BAL profiles.S5
Once established, airway inflammation in infancy may be associated with lower lung function and significant structural damage (including bronchiectasis) and poorer nutritional status.3 Neutrophils are the predominant inflammatory cells in CF airways and play a central role in the ensuing tissue damage and disease progression. These phagocytes possess an array of mediators, oxidants and proteases, including NE, that are critical for the neutrophil response against infection and bacterial killing, but large amounts of these enzymes escape from neutrophils in cell death and during phagocytosis. NE, a serine protease capable of digesting diverse substrates including structural proteins, is associated with lung function decline.4 Active NE injures the respiratory epithelium and weakens the airway structure resulting in bronchiectasis and bronchomalacia. The antiprotease defences in the airways such as secretory leucocyte protease inhibitors are designed to oppose free proteases and prevent their deleterious effects. However, these are eventually overwhelmed by the protease burden in the lung and are degraded by bacterial and human NE.S6 Several other neutrophil products such as defensins, active matrix metalloprotease-9 and calprotectin are found at elevated concentrations in respiratory secretions of patients with CF.5 Calprotectin, a proinflammatory protein present in large amounts in neutrophils, is found in high concentrations in both sputum and serum of patients with CF, and these levels decrease following treatment of an acute exacerbation.S7 In addition, CF airways are exposed to oxygen radicals derived from environmental oxygen and bacterial products and also from the host immune response. This oxidative stress exacerbates pulmonary deterioration and advances bronchiectasis in patients with CF. Similar to the protease/antiprotease balance, defence mechanisms of antioxidants are overwhelmed by the burden of oxidants in the CF airways.6
Extracellular triggers of CF airway inflammation
CF-airway cells are exposed to extracellular stimuli, such as infectious organisms, gastric acid, proteases and oxygen radicals which constantly incite an inflammatory response (figure 1).

Extracellular triggers of inflammation in cystic fibrosis (CF) airway cells. Various extracellular factors chronically present in CF airways continuously trigger the airway epithelial innate and adaptive immune cells to produce excessive proinflammatory cytokines.
To understand the relationship between infection and inflammation in the CF lung, the methods used to detect microbial pathogens need to be considered. Culture-independent methods for quantitative profiling of bacterial populations have opened a new era of investigation. While routine clinical and antibiotic therapies are largely directed by cultures, more sophisticated high-throughput molecular analysis techniques detect much higher bacterial diversity and complex fungal communities in the CF lung.7 However, the clinical significance of the lung microbiota is difficult to interpret.
Bacterial infection stimulates an intense neutrophilic response which fails to eradicate the infection leading to sustained release of proinflammatory mediators, chemoattractants and continuous influx of inflammatory cells.5 As the bacterial burden in the lung increases, the inflammatory response intensifies. In addition, evidence exists that inflammation in the CF airway is disproportionate to the infectious threat. CFTR-deficient mice have significantly higher concentrations of inflammatory mediators in BAL fluid and greater mortality than normal controls following intrabronchial instillation of Pseudomonas-laden agar beads.8 Epithelial sodium channel-overexpressing transgenic mice spontaneously develop features consistent with CF lung disease such as elevated levels of the CXC-chemokine macrophage inflammatory protein 2, tumour necrosis factor α (TNFα) and influx of neutrophils.S8 Further activation of the epithelial sodium channel by Pseudomonas aeruginosa may exacerbate this inflammatory response.S9 The lungs of newborn CF pigs, an animal model for CF-like lung disease, lack inflammation at birth but develop intense airway inflammation due to impaired bacterial clearance.S10
Data derived from primary murine tracheal cells suggest that, following P aeruginosa infection, inflammatory mediators are concentrated in the thin dehydrated periciliary fluid layer of CF airway epithelial cells resulting in high chemokine concentration gradients across the epithelium and an exaggerated inflammatory response.S11 In patients with CF, higher ratios of neutrophils or IL-8 to bacteriaS12 and reduced concentrations of the anti-inflammatory factor IL-10S13 were also found in BAL fluid compared with control subjects regardless of the pathogen recovered. The polymicrobial nature of airway infection including facultative anaerobic bacterial species in patients with CF may further exacerbate the inflammatory response, and inflammatory markers in BAL fluid increase with greater bacterial diversity.9 S14 However, the clinical significance of these bacteria in the progression of chronic pulmonary disease and during exacerbations in CF is yet to be determined.
Respiratory viral infections such as respiratory syncytial virus, influenza, parainfluenza, rhinovirus and adenovirus are deleterious to patients with CF leading to greater hospitalisation rates, growth impairment and reduction of lung function. Rhinovirus is a major contributor to pulmonary exacerbations in CF independent of bacterial infections.S15 Viral infections appear to predispose the CF airway to bacterial infection and correlate with earlier acquisition of P aeruginosa.S15 In addition, viral communities may contribute to CF pathology, altering the antibiotic resistance of resident microbes by transferring antibiotic resistance genes through phages.S16 Studies of CF airway epithelial cells have shown that planktonic bacteria from biofilm of mucoid Pseudomonas were released following rhinovirus infection, triggering a more robust inflammatory response, reduced apoptosis and increased viral replication.10 Thus, viruses contribute to airway inflammation in CF by directly infecting the airway and changing the antibiotic resistance patterns of bacterial flora which incite a greater inflammatory response.
Fungal infections may play a role in CF lung disease, although they are frequently underdiagnosed by routine laboratory cultures.7 The main clinically significant fungi recovered from respiratory secretions of patients with CF include Aspergillus spp, Candida spp, Scedosporium apiospermum and Exophiala dermatitidis. In a murine model, using CFTR-F508del mice, exposure to Aspergillus fumigatus hyphae caused profound lung inflammation and an enhanced Th2 inflammatory response characterised by elevated IL-4 in the lung.S17 Furthermore, inhaled hyphal forms induced secretion of inflammatory mediators including macrophage inflammatory protein 2, keratinocyte chemokine and monocyte chemotactic protein 1 and exaggerated lymphocytic inflammation, mucin accumulation and lung injury in CFTR-deficient mice.11 The respiratory disease in CF that is most often associated with fungal infection is allergic bronchopulmonary aspergillosis, which affects as many as 15% of patients with CF in different studies.S18 Additionally, Aspergillus sensitisation was associated with greater lung function decline and pulmonary exacerbations, suggesting damage secondary to the hypersensitivity response.S19
Severe gastro-oesophageal reflux and pulmonary microaspiration into the lung have been associated with chronic respiratory symptoms such as cough and wheeze and declining lung function in patients with CF.S16 Subjects with CF who had high levels of pepsin in BAL fluid or bile acids in sputum samples had higher IL-8 and NE concentrations,12 suggesting that aspiration of gastric contents is associated with more prominent airway inflammation. Animal studies have shown that repeated aspiration of acid into the tracheobronchial tree resulted in inflammatory cell infiltration to the airway and increased adherence of P aeruginosa to the airway epithelium.S20 S21 Gastro-oesophageal reflux has therefore been proposed as a precipitant for early inflammation in infants with CF, but this association still lacks hard evidence.
Reactive oxygen species-induced damage can be found even in infants and young children with CF, and the reactive oxygen species level in BAL fluid is associated with greater airway inflammationS22 and is inversely correlated with lung function. There is growing evidence that oxidative stress leads to oxidation of airway proteins, especially surfactant protein A (SP-A) and A1AT.S23 Glutathione, a ubiquitous tripeptide which exists in reduced monomeric (GSH) and oxidised dimeric (GSSG) forms, is a major component of cellular defence against oxidative injury and is present in large amounts in the epithelial lining fluid of the lower respiratory tract. Levels of reduced glutathione are depleted in the epithelial lining fluid and plasma from adult patients with CF.S24 Another possible mechanism for the depletion in reduced glutathione is lower GSH efflux found in CFTR-deficient cells, and depletion of reduced glutathione may further augment cytokine release and airway inflammation.13
Intracellular causes of excessive inflammation in the CF airways
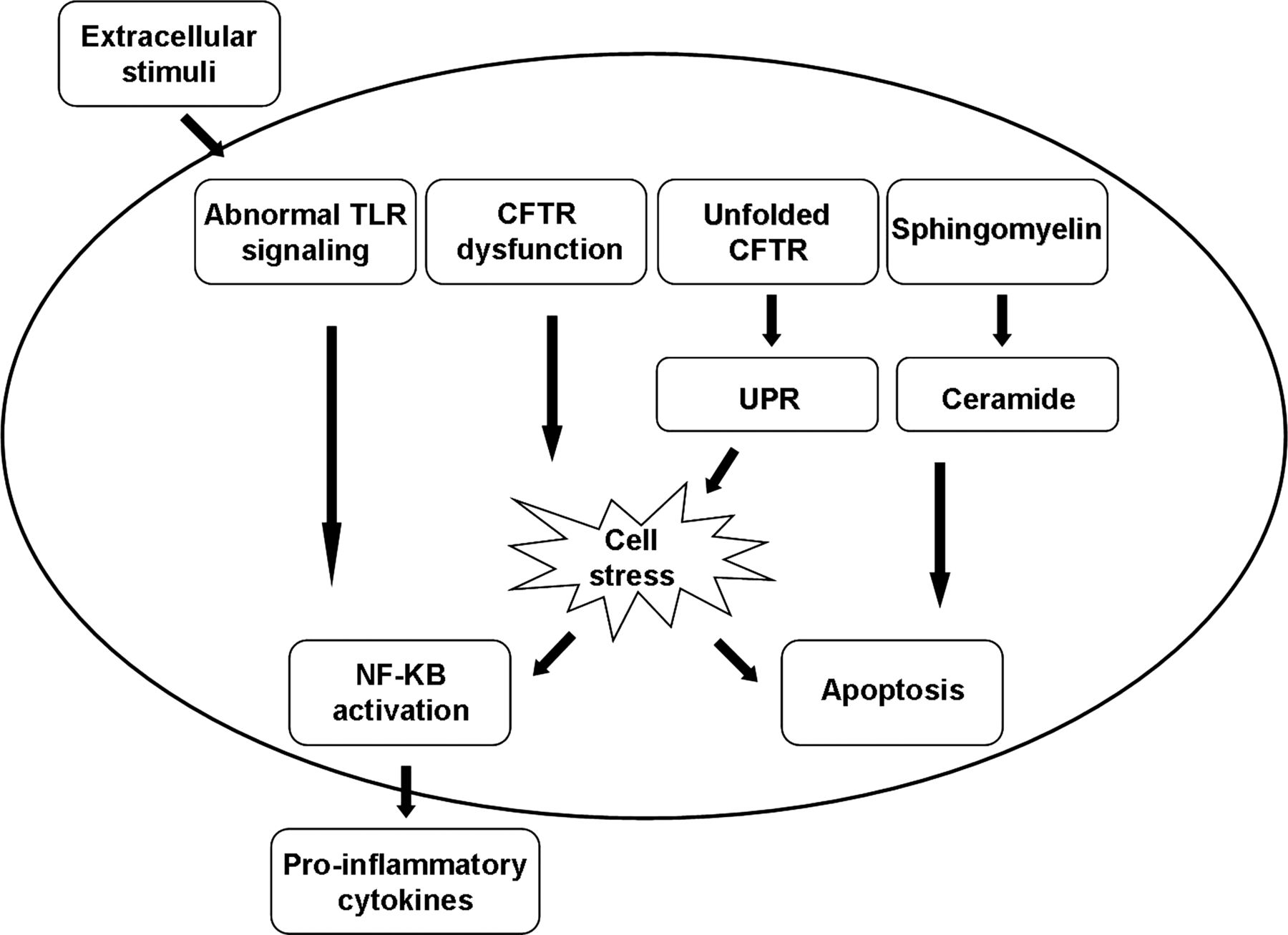
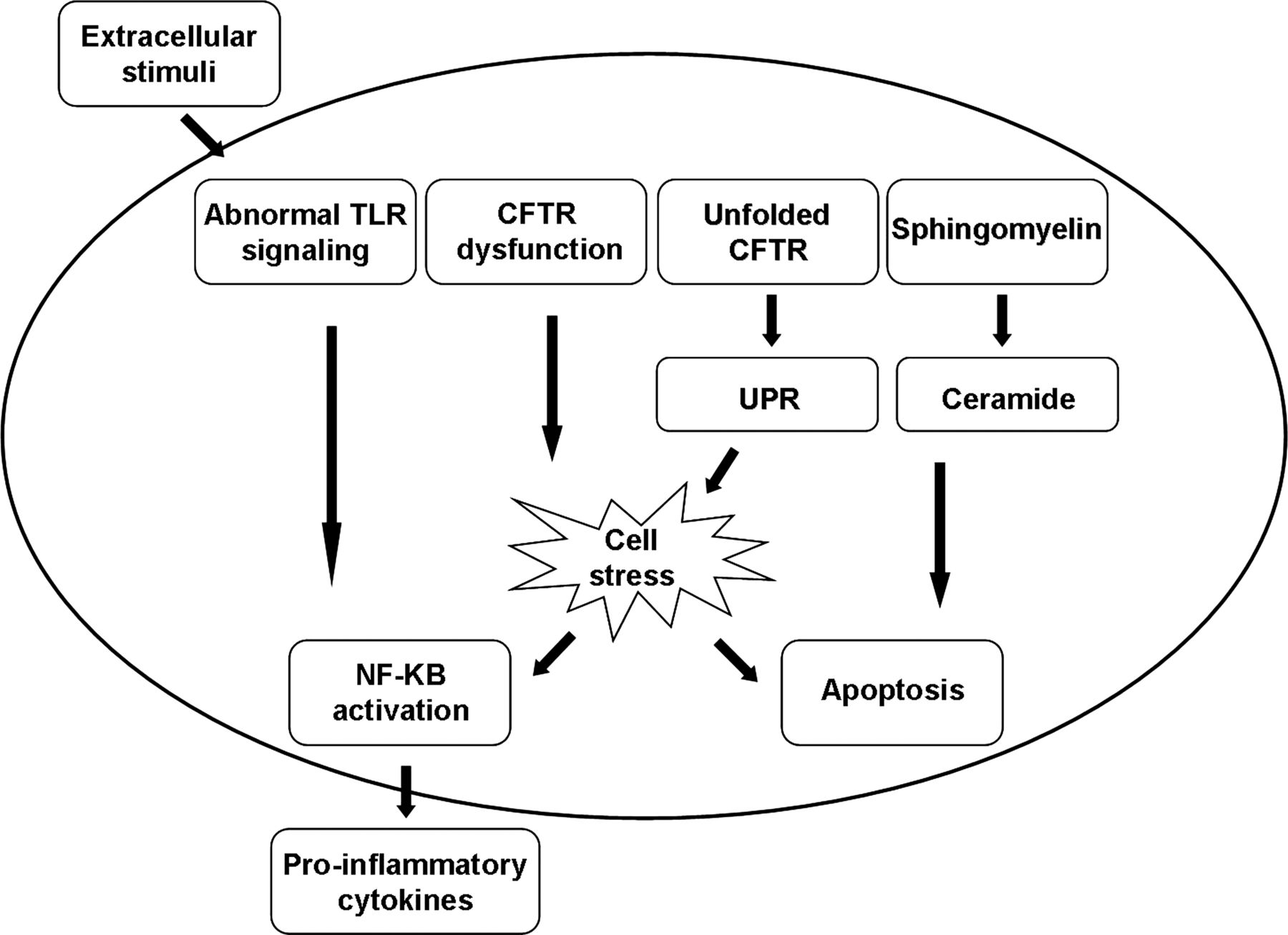
In CF, not only are the airway cells constantly exposed to extra-cellular pro-inflammatory triggers, but several lines of in vitro evidence also support the idea that the cellular responses to these triggers exceed the threat posed by the stimulus (see online supplementary table S1). However, not all studies have demonstrated an exaggerated inflammatory response in the CF airway. The apparent inconsistency of observed alterations in cellular responses to inflammatory stimuli in CF epithelial cells may be related to differences in cell models, stimuli and other experimental conditions, as shown in figure 2.14
{kind=link}
{kind=link}
Intracellular triggers of inflammation in cystic fibrosis (CF) airway cells. Inherent abnormal intracellular pathways, primarily in airway epithelial cells, lead to apoptosis on the one hand and to cell stress and excessive cytokine production on the other, perpetuating the inflammatory response in the airways. CFTR, CF transmembrane conductance regulator; TLR, Toll-like receptor; UPR, unfolded protein response.
Dysregulated activity of transcription factors has been suggested to underlie the proinflammatory properties of the CF airway epithelium. The p38 and ERK/MAPKs pathways are activated in some CF cell lines with a high abundance of classical NF-κB, p65 (RelA) and NF-κB, p65 (RelB) and inhibition of IκB kinase, the major inhibitor of NF-κB.S25 Levels of the lipopolysaccharide-inducible protein A20, an inhibitor of NF-κB signalling, are diminished in CF airway epithelial cells and its interactions with various intracellular proteins which facilitate its activity are altered.S26 Free NF-κB then translocates into the nucleus and increases the expression of proinflammatory genes in CF and non-CF cell lines. NF-κB activation has been associated with altered CFTR channel function. Inhibition of normal CFTR function in human tracheal epithelial cells resulted in NF-κB translocation and secretion of proinflammatory cytokines.S27
Ceramide is a breakdown product of sphingomyelin at various subcellular sites. Neutral sphingomyelinase hydrolyses sphingomyelin in the outer leaflet of the plasma membrane while an acidic sphingomyelinase produces ceramide in the endosomal-lysosomal compartments.S28 The ceramide formed is further degraded to sphingosine by acid ceramidase. It was suggested that, in CF, alkalinisation of intracellular lysosomal vesicles leads to ceramide accumulation in these cells. Ceramide accumulation may augment pulmonary inflammation by inducing apoptosis with subsequent deposition of DNA in the airways which increases adhesion of P aeruginosa.15 Susceptibility to P aeruginosa infections was shown in Cftr-deficient mice.S29 In addition, depending on the location and enzymatic pathway by which it is formed, ceramide is involved in distinct TNFα pathways. Downstream signalling events triggered by ceramide include the mitogen-activated protein kinase cascade and the induction of IκB-α degradation as well as nuclear translocation of NF-κB.S28 These independent processes, together with ceramide-induced IL-1 and CXC-chemokine upregulation, lead to chronic inflammation characterised by accumulation of neutrophils and macrophages in the lungs.S29
The response to accumulation of misfolded or unfolded proteins is another feature of the intrinsic proinflammatory CF phenotype. In eukaryotic cells, endoplasmic reticulum (ER) stress arises from the accumulation of misfolded or unfolded proteins in the ER, eliciting the unfolded protein response (UPR), an adaptive signalling pathway designed to resolve ER stress. UPR leads to a reduction of protein synthesis. When UPR signalling pathways are not sufficient to restore normal cell parameters, cells may activate the ER overload response leading to apoptosis.16 To repair tissue damage caused by ER stress-induced cell death, UPR mounts an inflammatory response. Genetic factors that determine the threshold of tolerable ER stress within the epithelium include genes that are centrally involved in the UPR, including X-box binding protein-1, anterior gradient protein-2 and orosomucoid-1-like 3.17 When ΔF508-CFTR is incorrectly folded and stuck in the ER, it induces UPR.S30 Retention of misfolded ΔF508-CFTR triggers ER stress and increases inflammatory signalling by endogenous activation of NF-κB.18 In addition, several inflammatory cytokines that stimulate apoptosis are induced by UPR. However, while this phenomenon may account for a robust inflammatory response in class II CFTR mutations, it does not explain inflammation associated with other CFTR mutations.
The initial host defence to the loss of CFTR leads to intracellular alkalinisation and pH reduction in the airway surface fluid layer. Antimicrobial function is inhibited, impairing the killing of bacteria in the lung which was recently demonstrated in the CF pig model, indicating that increasing the pH of the airway surface fluid layer might prevent the initial infection in CF piglets.19
Dysregulated innate and adaptive immune responses were also described in subjects with CF (see online supplementary table S1). Fetal CF lungs had distinct populations of mast cells and macrophages compared with non-CF fetuses,S31 which suggested that early alterations in immune cell distribution may contribute to the early onset of inflammation. Further support for the dysregulation of the innate immunity in CF stems from recent studies demonstrating that alveolar macrophages and related CC-chemokines were elevated in BAL fluid from patients with CF, even in the absence of infection,20 and that the expression of Toll-like receptor 4 (TLR-4), a pattern recognition factor of bacterial lipopolysaccharide on peripheral blood monocytes was enhanced in patients with CF, unrelated to the presence (or absence) of pulmonary infection.S32
The function of other cells of the innate immune system may be dysregulated as well. Neutrophils preferentially accumulate in the surface CF epithelium, possibly due to overexpression of intercellular adhesion molecule-1.S33 The cells recruited to the CF airways exhibit a viable phenotype expressing surface markers that are usually associated with other cell lineages.S34 They were also found to increase TLR-4 expression on their surface and produce excessive proinflammatory cytokines at baseline, while failing to increase secretion in response to lipopolysaccharide or repress it in response to IL-10.21
Cells of the adaptive immune response such as T lymphocytes may also be involved in the immune dysregulation in patients with CF. T cells are more abundant in CF airways and spatially associated with greater tissue damage.S33 Circulating T lymphocytes were shown to produce low levels of IL-8 and, by the emergence of more numerous T cells, to produce high levels of IL-2. This imbalance may contribute to immune dysregulation in CF.22 T cells may also play a significant role in the production of IL-17, a cytokine regulating neutrophil recruitment. High levels of IL-17 and IL-23, a regulator of Th17 production, were found in BAL fluid of adult patients with CF during pulmonary exacerbations,S35 and Th17 lymphocytes are present in the submucosa of endobronchial biopsies from children with CF, even early in the course of the disease. Increased numbers of IL-17 (+) cells were found even in children with newly diagnosed CF who had BAL neutrophilia in the absence of elevated IL-8, suggesting that IL-17 cells are important early in CF lung disease.23
Assessing inflammation in the CF airways
Measuring airway inflammation in CF is important for initiating anti-inflammatory treatment and monitoring its effect (table 1). At present, no inflammatory biomarker has been consistently shown to predict clinical efficacy. Forced expiratory volume in one second (FEV1), a surrogate to assess severity of CF lung disease, has become the gold standard for monitoring disease progression. In some but not all studies, FEV1 inversely correlated with total cell count and free NE in BAL fluid obtained from patients with CF.2 Due to improved CF care in recent decades, the decline in FEV1 has slowed, reducing its impact as a disease predictor. Thus, prolonged follow-up may be required before an effect of anti-inflammatory therapy can be detected, further limiting the value of FEV1 as an indirect surrogate marker for inflammation in CF.
Methods of assessing inflammation in the cystic fibrosis airways: pros and cons
Ventilation inhomogeneity, measured by multiple breath washout and expressed as the lung clearance index (LCI), has emerged as a valuable tool for measuring small airway function in patients with CF with normal or mildly reduced FEV1. LCI was associated with structural abnormalities in the small airways and with inflammatory markers such as IL-8 and neutrophil count in BAL fluid, and was recently used as a clinical trial endpoint for patients with CF with mild pulmonary involvement.24 S36 Nevertheless, large longitudinal studies are still needed to verify its prognostic value.
Sampling the lower respiratory secretions through BAL is another approach to assess CF airway inflammation.24 However, its results may not be reproducible, probably due to the heterogeneity of airway inflammation in CF. In addition, the invasive nature of this technique limits its use. Induced sputum has safety advantages over BAL. However, while some studies have shown an association between inflammatory markers, especially NE, in sputum from patients with CF and lung function decline,4 others have found no correlation between sputum cytokine concentrations and clinical disease severity. There is still much controversy regarding the reproducibility of this approach to monitoring airway inflammation in CF.S37
Endobronchial biopsy can detect early pathological changes in the CF lung. Airway remodelling, defined as structural airway wall changes, begins early in life in CF. An increased basement membrane thickness that correlated with BAL fluid concentrations of transforming growth factor-α1 was demonstrated in children with CF.25 In adults with advanced CF disease this approach revealed epithelial squamous metaplasia, loss of ciliated epithelial cells and ultrastructural abnormalities such as disorganisation of tight junctions and compound cilia. More recently, accumulation of lymphocytes was shown in bronchial mucosa, in contrast to neutrophil-dominated inflammation present in the airway lumen.26 Although endobronchial biopsies have been advocated as a tool to evaluate airway inflammation and remodelling in CF,27 S38 this procedure is highly invasive and cannot be used routinely to monitor airway inflammation.
Analysis of exhaled breath condensate (EBC) for various inflammatory markers could provide a non-invasive assessment of CF airway inflammation. Carbon monoxide and volatile hydrocarbons are elevated in EBC of patients with CF and increase during pulmonary exacerbations. Other inflammatory markers, including leukotriene B4, IL-6 and IL-8, were also shown to be elevated in EBC from patients with CF. EBC pH reflects the acid-base homeostasis of the airway lining fluid and is lower in various inflammatory lung diseases including CF.28 However, the usefulness of EBC to monitor CF airway inflammation still needs to be established. Fractional exhaled nitric oxide (FeNO), a non-invasive marker for measuring inflammation, was reported to be low in patients with CF and values are independent of the degree of inflammation. Furthermore, no correlation was found with pulmonary exacerbations and antibiotic therapy.29
High-resolution CT (HRCT) of the chest can show structural changes that indirectly reflect the degree of inflammation in the CF lung, correlating with levels of sputum inflammatory markers such as IL-8 and TNFα.S39 However, these changes may be the result of past airway injury and do not directly reflect active inflammation. Thus, its role as a surrogate outcome measure for clinical trials evaluating the efficacy of anti-inflammatory drugs is still unclear. Furthermore, despite new low-radiation protocols, its safety and applicability for regular monitoring are still major concerns.
Given the invasiveness of many of the aforementioned techniques, serum biomarkers recently gained renewed interest as a means of assessing airway inflammation and response to treatment. In a recent study examining the effect of azithromycin on systemic markers of inflammation in children with CF, calprotectin was associated with FEV1.S40 Other investigators have reported a decline in various serum markers of inflammation including C reactive protein, IL-6 and calprotectin following treatment of pulmonary exacerbation in patients aged >10 years. These parameters also correlated with symptoms score, LCI and several imaging findings.S41
Finally, an innovative outcome measure is positron emission tomography (PET) using 18fluordeoxyglucose (18FDG PET) which exploits the rapid uptake of 18FDG into neutrophils by metabolic trapping. The 18FDG PET signal was shown to correlate with neutrophilic inflammationS42 and could be used to determine the location and intensity of 18FDG uptake in the lungs. Current technology allows for simultaneous CT imaging for anatomical localisation of PET signals.30 S42 Cross-sectional data from adults with CF have shown that 18FDG uptake correlates with BAL fluid neutrophil counts and can detect an effect of intravenous antibiotic therapy for a pulmonary exacerbation in CF.30S42 Despite these promising results, radiation is a concern which restricts its routine use for clinical indications.
Future prospects
The progress in understanding the relationship between CF airway inflammation and infection may lead to new directions in treatment. The currently available animal models that mimic human CF lung disease should provide important insights into the origins of airway inflammation.31 S10 S43 Genetic modifiers could increase or attenuate inflammation in the CF lung, and newer powerful technologies provide an attractive approach to identify modifier loci of Mendelian disorders. In genome-wide single nucleotide polymorphism association studies (GWAS) combining data from three different study populations, by minimising heterogeneity using mostly patients homozygous for ΔF508-CFTR mutations and advanced lung disease, modifier genes were identified including APIP (Apaf-1-interacting protein) which inhibits apoptosis. Decreased neutrophil apoptosis could translate into delayed resolution of inflammation and lead to airway damage and deteriorating pulmonary function.32 In another GWAS study an association was found between the interferon-related developmental regulator-1 (IFRD1) expressed late in neutrophil differentiation and severity of CF lung disease. Neutrophils from IFRD1-deficient mice showed blunted effector function associated with decreased NF-κB p65 transactivation.S44
Induced pluripotent stem cells (iPSCs) are a novel tool for CF research. Recently, murine lung and airway progenitors were generated and disease-specific lung progenitor cells from human CF iPSC were shown to form respiratory epithelium when subcutaneously engrafted into immunodeficient mice.33 In another study functional CFTR-expressing airway epithelia were generated from human embryonic stem cells. These initial studies may provide a new platform for studying the development of CF lung disease and its relationship with inflammation.34
Conclusion
Inflammation develops in CF airways early in life in response to extracellular infectious stimuli and possibly due to immune dysregulation which includes abnormal TLR signalling, unfolded CFTR stress response and abnormal innate and adaptive immunity. The failure of the immune response to eradicate infectious organisms from CF airways has received much attention in recent years, but the self-perpetuating inflammatory response accounts for much of the lung pathology seen in patients. Newer animal models which mimic human CF lung disease and other technologies may improve our understanding of lung inflammation in CF and help in the development of new therapies in future years. In the meantime, better methods to measure inflammation in the CF airways need to be developed, which are crucial for treatment with and monitoring of anti-inflammatory agents.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors MC-C and AE made substantial contributions to the conception and design of the study and drafting of the article. EK and TF revised the article critically for important intellectual content. MC-C, EK, TF and AE approved the final version to be published.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.