Article Text

Abstract
Background Molecular methods based on phylogenetic differences in the 16S rRNA gene are able to characterise the microbiota of the respiratory tract in health and disease.
Objectives Our goals were (1) to characterise bacterial communities in lower and upper airways of patients with interstitial lung disease (ILD) and (2) to compare the results with the microbiota of patients with Pneumocystis pneumonia (PCP) and normal controls.
Methods We examined the upper and lower respiratory tract of 18 patients with ILD of whom 5, 6, and 7 had idiopathic interstitial pneumonia (IIP), non-IIP and sarcoidosis, respectively. In addition, six immune-compromised patients with PCP and nine healthy subjects were included as controls. Exclusion criteria were recent bacterial/viral respiratory tract infection, HIV-positivity and subjects receiving antibiotic therapy. Bronchoalveolar lavage fluid and oropharyngeal swabs were simultaneously collected, and microbiota was characterised by ultra-deep 16S rRNA gene sequencing.
Results The microbiota in lower airways of the majority of patients (30; 90%) primarily consisted of Prevotellaceae, Streptococcaceae and Acidaminococcaceae. α and β diversity measurements revealed no significant differences in airway microbiota composition between the five different groups of patients. Comparison of bacterial populations in upper and lower respiratory tract showed significant topographical discontinuities for 7 (23%) individuals.
Conclusions IIP, non-IIP and sarcoidosis are not associated with disordered airway microbiota and a pathogenic role of commensals in the disease process is therefore unlikely. Nevertheless, molecular analysis of the topographical microbiota continuity along the respiratory tract may provide additional information to assist management of individual patients.
- Sarcoidosis
- Immunodeficiency
- Bacterial Infection
- Respiratory Infection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is the upper and lower airway microbiota of patients with interstitial lung disease disordered?
What is the bottom line?
-
For the majority of patients with interstitial lung disease, the lung microbiota is comparable with normal controls.
Why read on?
-
Using molecular methods, we describe the microbiota of the upper and lower airways in health and disease, which has the potential to improve our understanding in the underlying causes of lung diseases as compared with conventional culture alone.
Introduction
Lower airways in healthy individuals have been considered as being sterile based on conventional microbial culture methods.1 ,2 This paradigm has recently been challenged by studies implementing ultra-deep sequencing methods analysing the 16S rRNA gene. In particular, it has been suggested that lower airways might be colonised with ‘symbiotic’ bacteria.3 ,4 Furthermore, it has been revealed that a disturbance in the composition of the lower airway microbiota may be associated with chronic lung diseases, such as asthma or chronic obstructive lung disease.3 ,5–8
A disturbance of the microbiota may also be present in patients with interstitial lung disease (ILD), but so far, this has not yet been investigated. Though it is known that immune-compromised hosts are at increased risk for lower respiratory tract infections, the distinction between non-infectious and infectious causes for deterioration of an ILD patient can be clinically challenging based on conventional microbiological diagnostic techniques.9 An alteration of the airway microbiota in ILD patients may hypothetically be associated with clinical worsening due to immune dysregulation, excessive inflammation and/or infection. The characterisation of microbiota may therefore be of an additional diagnostic value.
In the underlying study, we sought to investigate the bacterial microbiota in the upper and lower respiratory tract in patients with ILD. More specifically, we aimed at addressing two objectives: (1) to characterise bacterial communities in lower and upper airways of patients with ILD and (2) to compare the results with the microbiota of patients with Pneumocystis pneumonia (PCP) and normal controls.
Material and methods
Study population and definitions
The Epidemiology and Clinical Relevance of Infectious Agents in Bronchoalveolar Lavage (ECRIBAL) study is a prospective, single centre observational study investigating new diagnostic methods and potential pathogens in patients with ILD and in immune-compromised patients with suspicion of lung infection. The study has been approved by the local ethics committee (Kantonale Ethikkommission Bern, approval Nr. 153/08).
For the purpose of the current microbiota study and based on the ATS/ERS international multidisciplinary consensus classification,10 we included patients of the following three ILD groups from ECRIBAL: exacerbated idiopathic interstitial pneumonias (IIP) (n=5), exacerbated non-IIP (n=6) and ILD due to sarcoidosis (n=7). Based on this classification, the non-IIP group contained ILD due to known causes, such as collagen vascular disease, environmental, drug related and ‘other’ ILD, for example, eosinophilic pneumonia (see table 1 for details). In addition, we included the following comparative groups: patients with Pneumocystis pneumonia (PCP) (n=6) and healthy controls without evidence for pulmonary or systemic disease (n=9).
ILD (IIP, non-IIP, sarcoidosis), PCP and control subgroups used for analysis
Enrolled individuals with an indication for bronchoscopy at Bern University Hospital (Switzerland) had to be ≥18 years of age and able to provide informed consent. Exclusion criteria for this study were: recent bacterial/viral respiratory tract infection within 2 weeks prior to bronchoalveolar lavage (BAL), HIV-positivity and subjects that had received antibiotic therapy within 48 h prior to BAL as this was previously shown to affect the airway microbiota.11 ,12 The time difference between BAL and lung function was normally 2 weeks. We initially selected 41 individuals out of whom 8 individuals from the following groups had to be excluded due to absent amplification of lower airway microbiota from BAL: sarcoidosis (n=3), non-IIP (n=3) and controls (n=3).
BAL procedure and microbiological analysis
BAL was performed during bronchoscopy by wedging of the bronchoscope tip in a segmental bronchus of the lobe that displayed radiological changes, followed by fractional instillation and withdrawal of a total of 150 mL prewarmed saline using 50 mL syringes. Microbiological investigations in BAL consisted of routine bacterial, fungal, viral and mycobacterial culture and a panel of 15 respiratory viruses routinely detected (antigen detection and/or PCR).13 Additional immunofluorescence and PCR detection of P. jirovecii were performed as previously described.14 Finally, oropharyngeal (OP) swabs were taken for subsequent analysis of viruses and the microbiota of the upper respiratory tract.
PCR amplification of the 16S rRNA genes
DNA extraction was performed using 200 μL of BAL sample and the OP swab as previously described.15 ,16 V3–V5 regions of bacterial 16S rRNA genes were amplified using the primer pairs 341F/926R.17 Primer sequences (bold) were modified by addition of the Roche 454 Titanium sequencing A or B adaptor sequence (lower cases) and a 10-mer multiplex identifier (A-MID-341F, 5′-cgtatcgcctccctcgcgccatcag-[NNNNNNNNNN]-ACTCCTACGGGAGGCAGCAG-3′; B-MID-926R, 5′-ctatgcgccttgccagcccgctcag-[NNNNNNNNNN]-CCGTCAATTCMTTTGAGTTT-3′. PCR reactions were performed and purified using Wizard SV PCR clean-up system (Promega, Madison) as described.17 The final elution step was performed using 40 µL of double distilled water.
454 Titanium amplicon sequencing
Samples were pooled using 10 ng of PCR product of each sample resulting in eight different pools including every multiplex identifier (MID) once. The amplicon libraries were sequenced according to the 454 Titanium Amplicon Sequencing protocols and a series of quality control steps were applied to the resulting 454 reads.17 454 raw reads were submitted to the NCBI Sequence Read Archive under the sequencing experiment SRA026964 with the accession numbers SRX033134 and SRX193716 (normal control samples).
Calculation of richness and Shannon diversity indices and community comparisons (α and β diversity)
PyroTagger was used for the definition of operational taxonomic units based on 97% sequence identity, estimation of chimaeras and taxonomy assignments.18 α diversity analysis (including richness, Shannon and Simpson Diversity indices) was performed employing Mothur.19 For the statistical analysis of the α diversity results, ordinary one-way ANOVA and Mann–Whitney analyses were performed. Resulting graphs were generated with GraphPad Prism V.5.02 (GraphPad Software, Inc, La Jolla, California, USA).
Community comparisons were calculated using non-metric multidimensional scaling (nMDS) and the adonis function of the vegan package in R as previously described.17 For the creation of the input distance matrices, binary and abundance-based (Horn) calculations were performed as previously described.17 ,20 Procrustes function of R was used to compare the nMDS plots between the upper and lower respiratory tract.
Ordinary one-way ANOVA was performed to investigate the significant differences of the relative mean abundances of bacterial families of the upper and lower airways.
Results
Study patients, PCR amplification and 16S rRNA gene pyrosequencing
Clinical characteristics of the 33 patients are summarised in table 1. The study subjects had an overall mean age of 55.2±13.7 years and were categorised into the following groups: IIP, non-IIP, sarcoidosis, PCP and healthy controls. Nine (27.3%) were women, sarcoidosis was the most frequent ILD diagnosis (n=7) and none of the 33 study subjects received antibiotic treatment within 48 h before BAL. Though amplification of the 33 BAL samples revealed that the bacterial quantity of controls was lower, this was not statistically significant. Subsequent 16S rRNA gene pyrosequencing was performed and estimates revealed that sequencing depth was approximately 80% (data not shown).
α Diversity measurements in lower airways
In order to investigate if microbiota composition varied between patients with different manifestations of ILD, we initially calculated bacterial richness and Shannon diversity indices of the lower airways, that is, in samples obtained from BAL (α diversity; figure 1). Both indices are ideal measurements to easily and rapidly investigate alterations in the microbiota as they result in a numeric value that may serve as a surrogate marker for disordered microbial communities. This value decreases when a single bacterial species has ‘overgrown’ and become predominant, replacing the classic ‘commensal’ microbiota.

α-Diversity indices of the lower airway microbiota. Indicated are the Richness (A) and Shannon-diversity (B) indices for subjects with idiopathic interstitial pneumonia (IIP), non-idiopathic interstitial pneumonia (non-IIP), sarcoidosis, Pneumocystis pneumonia (PCP) and normal controls. No significant differences were observed.
The mean richness values were 56.2 (±29.2), 83.7 (±20.6), 75.1 (±25.9), 60.3 (±16.4) and 63.3 (±20.3) for IIP, non-IIP, sarcoidosis, PCP and normal controls, respectively (figure 1A). The lowest value was therefore found for IIP but this finding was not statistically significant compared with the other groups.
As for the Shannon diversity indices, again, patients with IIP had the lowest values (mean 2.0±1.1) (figure 1B), but differences among the indices were again not significantly different performing both ordinary one-way ANOVA and Mann–Whitney analyses (figure 1A).
β Diversity measurements in lower airways
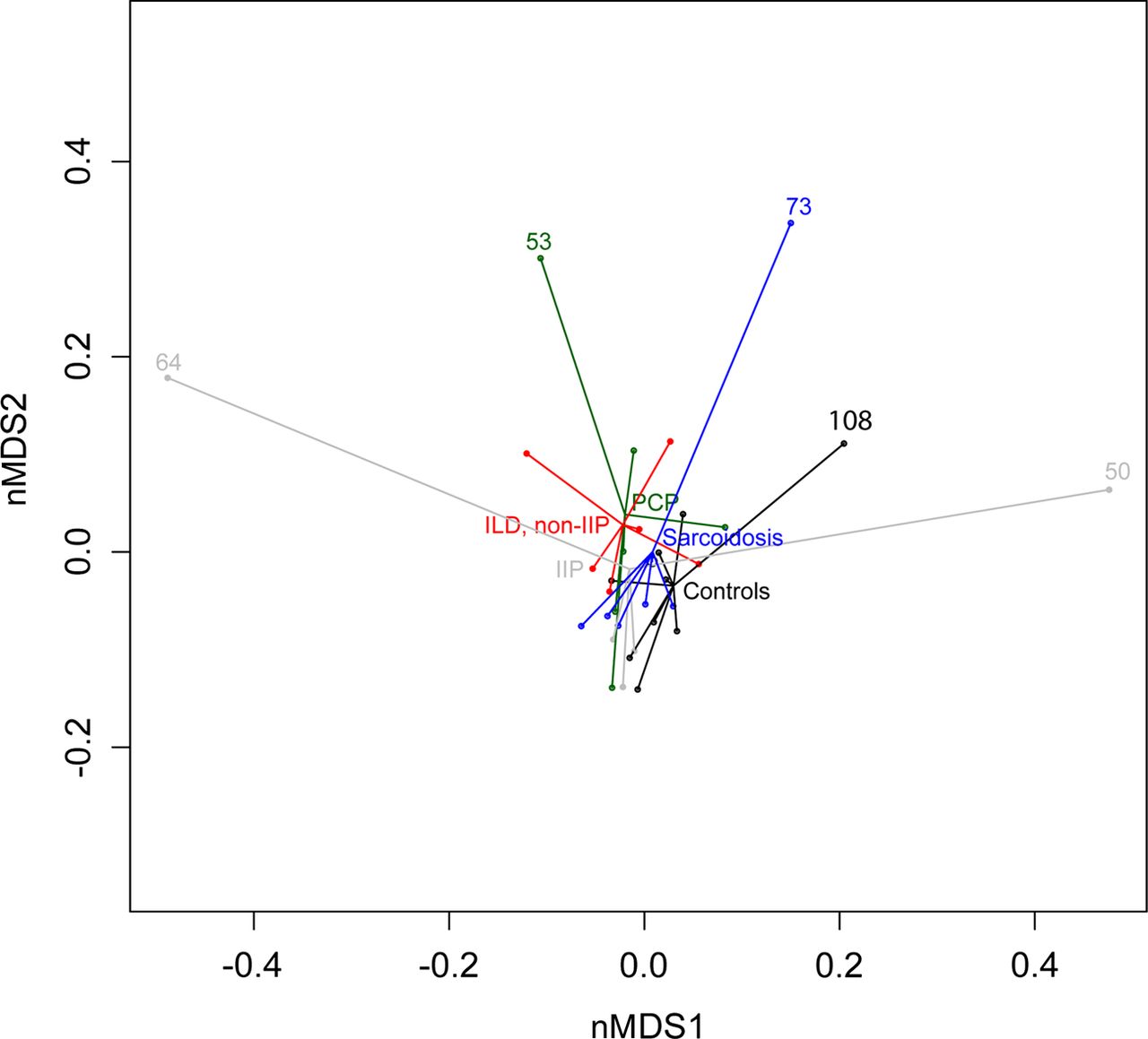
We then measured pairwise distances between microbial communities of all 33 samples. For this, Bray–Curtis distance matrices were used as input and the amount of variance was calculated (β diversity calculations). Overall bacterial communities in lower airways did not significantly differ in patients with IIP, non-IIP, sarcoidosis, PCP and controls (figure 2). However, five patients had a clearly different bacterial profile (ID 64, 53, 73, 108 and 50; figure 2).

Comparison of bacterial communities in the lower airways by a non-metric multidimensional scaling ordination plot. Shown are the results for subjects with idiopathic interstitial pneumonia (IIP), non-idiopathic interstitial pneumonia (non-IIP), sarcoidosis, Pneumocystis pneumonia (PCP) and normal controls. Bray–Curtis distance matrices were used as input. Statistical differences between the five groups were non-significant (ie, small distances between central points). Five patients had different microbiota illustrated by the large distances to the central points (ID 64, 53, 73, 108 and 50). nMDS, non-metric multidimensional scaling.
Relative abundance of bacterial families and quantification
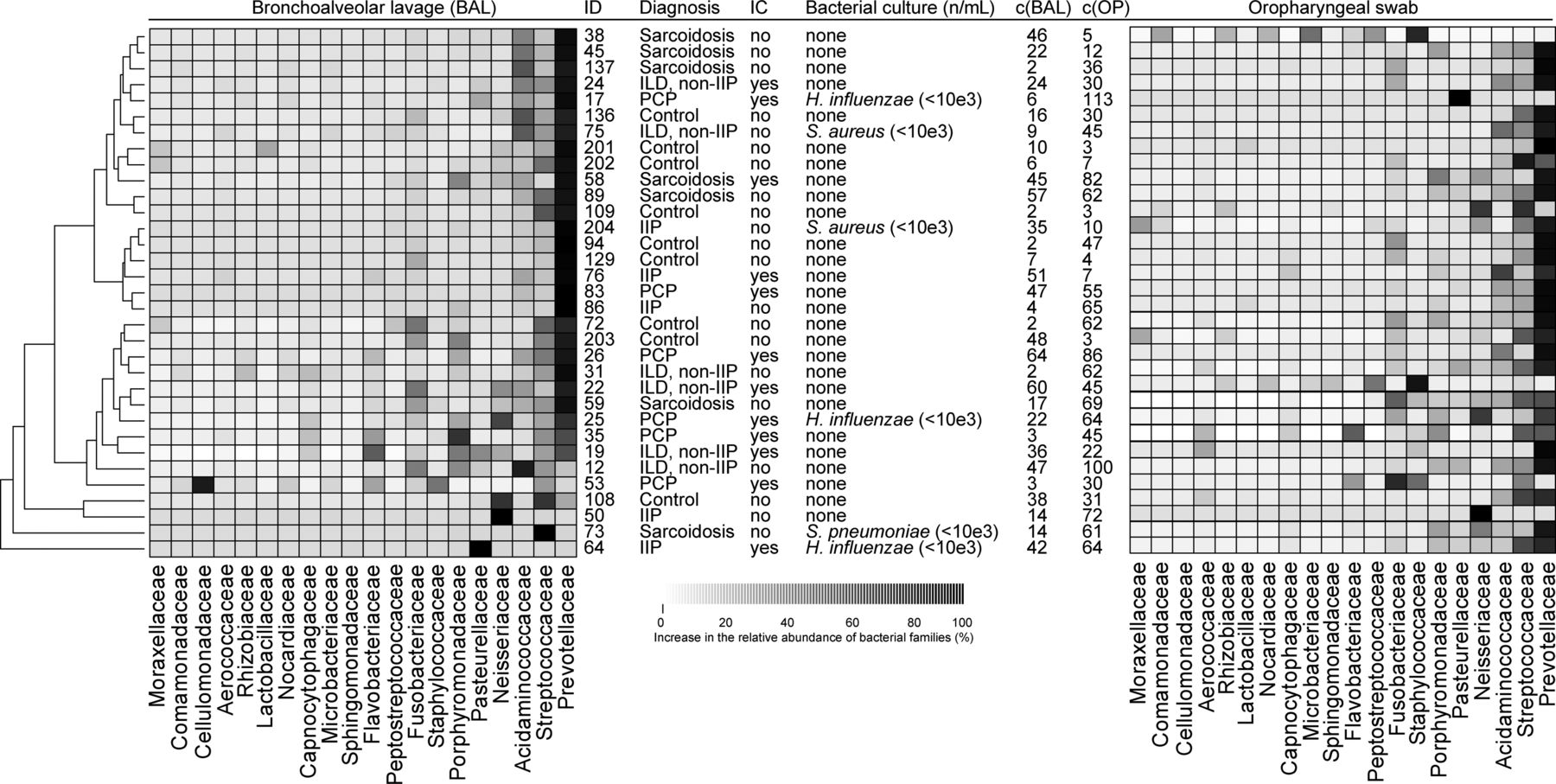
Sequencing results and subsequent taxa assignments for all 33 BAL samples are illustrated in figure 3. The communities of three patients harboured solitary abundances of Neisseriaceae (patient ID 50), Streptococcaceae (patient ID 73) and Pasteurellaceae (patient ID 64) (figure 3). However, the microbiota of the 30 remaining patients primarily consisted of bacterial families known as commensals, that is, non-pathogenic bacteria colonising the airways. Such commensals included Prevotellaceae, Streptococcaceae and Acidaminococcaceae. Due to the nature of the bacterial families present, this again indicated that the microbiota may not be disturbed in IIP, non-IIP, sarcoidosis and PCP as compared with healthy controls. In addition, a more detailed investigation of the relative differences of the bacterial families revealed that the clinical diagnosis (IIP, non-IIP, sarcoidosis, PCP or controls) is only significantly associated with the relative mean abundance of Flavobacteriaceae (table 2). For all other families, no significant differences were detected (table 2).
Relative mean abundances of bacterial families in lower and upper airways

Microbial community comparisons and relative abundances of the top 20 bacterial families in 33 bronchoalveolar lavage (BAL) and oropharyngeal samples. A tree illustrating similarities of the microbial communities of the BAL samples are shown on the left. Abundances of bacterial families for each subject (ID) are indicated by a greyscale value. The five groups consisting of idiopathic interstitial pneumonia (IIP), non-idiopathic interstitial pneumonia (non-IIP), sarcoidosis, Pneumocystis pneumonia (PCP) and normal controls, as well as if immunocompromised (IC) for each study subject are indicated. The quantities of bacterial species derived from conventional culture are provided in colony forming units (CFU). The concentrations of the amplification products (c) are indicated in ng/μL.
Analysis of topographical continuity along the respiratory tract
We subsequently analysed the upper respiratory tract (oropharynx) of the 33 subjects (figure 3; right column). As for the lower airways, Prevotellaceae was the most abundant bacterial family followed by Streptococcaceae and Acidaminococcaceae.
In a next step, we quantified differences between upper and lower respiratory tract communities in order to analyse topographical continuity. Assuming that a normal respiratory tract has similar or concordant microbiota profiles between upper and lower airways,4 a divergence would be indicative of the presence of disordered microbial communities at the site of interest. Using procrustes analysis we revealed that 7 of 33 patients (ID 17, 38, 50, 53, 64, 73 and 108) displayed differences exceeding the upper quantile of procrustes errors, suggesting a significant disordered microbiota between the upper and lower respiratory tract (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of the microbiota between upper and lower airways by procrustes analysis. Pairwise distance matrices were calculated between lower and upper microbial respiratory tract communities from which non-metric dimension plots were subsequently derived. Patients with procrustes residual exceeding the upper quantile (upper dashed line) were considered as containing different microbial communities (ID 64, 50, 73, 38, 53, 17 and 108). Study subjects are presented according to the clinical diagnosis: sarcoidosis, idiopathic interstitial pneumonia (IIP), non-idiopathic interstitial pneumonia (non-IIP), Pneumocystis pneumonia (PCP) and normal controls. ILD, interstitial lung disease.
We then analysed clinical characteristics of the seven patients with topographical microbiota discontinuity. Patient 38 had mainly commensal Corynebacteriaceae in the oropharynx, but essentially harbouring ‘normal’ Prevotellaceae, Acidaminococcaceae and Streptococcaceae in the BAL. Therefore, this subject could be considered as healthy in the absence of clinical signs and symptoms indicative of an upper respiratory tract infection. The high procrustes residuals in patients 17 and 64 have their origins in the high abundances of Pasteurellaceae (ie, Haemophilus influenzae as confirmed by culture) in the upper (patient 17) and lower (patient 64) respiratory tract (figure 3). H. influenzae is frequently detected in microbial cultures, but it is often unclear if this reflects non-pathogenic colonisation or infection, given that different strains with both features are known.21 In our study, we identified H. influenzae in patients 17, 25 and 64, but only in patient 64, the overall microbiota was disordered in the lower respiratory tract, suggesting infection rather than colonisation (figure 3). Patient 64 was diagnosed with idiopathic pulmonary fibrosis without evidence by conventional microbiology for respiratory tract infection and did not improve under an initial course of systemic corticosteroid treatment. Based on the microbiota results of the lower airways, H. influenzae may have played a clinically relevant role in the course of the exacerbation, and therefore, in the absence of the ‘normal’ microbiota, an initial antibiotic treatment may have been an appropriate treatment option.
The remaining procrustes differences occurred in patients 50, 53, 73 and 108. These were based on excess abundance of Streptococcus pneumoniae (patient 73), Neisseriaceae (patient 50 and normal control 108) and Cellulomonadaceae (mainly Tropheryma whipplei, patient 53) and were localised to the lower airways. In patient 73 with a histologically proven sarcoidosis, microbiota discontinuity was present with an excess of S. pneumoniae, but without signs for respiratory tract infection. This finding, together with the excess Neisseriaceae in patients 50 and 108, remains unclear. Finally, identification of Cellulomonadaceae (mainly T. whipplei) in patient 53 is an intriguing finding and may be due to hitherto undiagnosed Whipple's Disease.
Discussion
Recent studies using 16S rRNA gene pyrosequencing techniques have explored the microbiota of the lower and upper respiratory tract in health and disease.3 ,4 ,22 ,23 These techniques now also enable to investigate disordered microbial communities in patients with other lung disorders and conditions, such as IIP, non-IIP and sarcoidosis, which to our knowledge, has not been performed to date. Within this study, we therefore aimed at characterising the bacterial communities in lower and upper airways of patients with IIP, non-IIP and sarcoidosis and to compare the results with the microbiota of patients with Pneumocystis pneumonia (PCP) and normal controls.
Most of the patients with IIP, non-IIP, sarcoidosis and PCP harboured three bacterial families (Prevotellaceae, Streptococcaceae and Acidaminococcaceae) in the respiratory tract that were also frequently detected in healthy controls, and therefore, indicated that no disordering of the microbiota was present. It is worthwhile mentioning that the same commensal families have also been reported in other recent studies analysing the healthy respiratory tract.3 ,4
α and β diversity measurements are available to investigate if clinical factors and/or diagnoses are significantly associated with an altered microbiota composition. In our study, neither α nor β diversity indices revealed any significant changes related to the clinical diagnosis of IIP, non-IIP, sarcoidosis or PCP compared with healthy controls.
To date, sound data linking microbiota changes to lung disorders are still scarce as this field of research is still in its infancy. In HIV-infected patients, several known or suspected pathogenic organisms have been detected that may be related to recurrent pneumonia.11 In patients with non-CF bronchiectasis, characteristics of the lower airways microbiota correlated with clinical markers of disease severity.23 Finally, two studies revealed that the microbiota in cystic fibrosis patients is distinct, probably due to the specific environmental characteristics in the respiratory tract of these patients.24 ,25
In our study, we simultaneously analysed upper and lower respiratory tract, which is an important aspect for two reasons: first, this approach allows the examination for possible contamination from the upper respiratory tract.26 Second, it enables to specifically localise microbial communities in the airways as shown by recent studies.5 ,27 For the healthy human respiratory tract, topographical continuity of bacterial populations has previously been described.4 Within our study, we now extend these findings by demonstrating that continuity of microbiota also exists in the majority of patients with underlying ILD. This finding was not applicable for seven individuals revealing significant divergence between the upper and lower respiratory tract. This may indicate disturbed microbiota in the upper or lower airways, but has to be investigated for each patient separately. Based on our data, we suggest that the identification of microbiota discontinuity along the respiratory tract should generally be interpreted by taking into account the clinical presentation of the patient.
Compared with conventional bacterial cultures, utilisation of pyrosequencing methods generally has two advantages. First, the methods are able to identify unusual pathogenic bacteria normally not detectable by conventional microbiological methods.28–30 Second, these methods can assist to identify the pathogenic role of ‘commensal’ bacteria. Depletion of the normal microbiota with ‘overgrowth’ of a single bacterial species may indicate that this is clinically relevant, for example, as seen with H. influenzae in our study. This is an important aspect as the clinician is commonly confronted with the challenge to distinguish between innocuous commensals, ‘facultative pathogenic’ and ‘pathogenic’ bacteria. However, it is crucial to emphasise that conventional culture and molecular methods should be seen as complimentary, rather than exclusionary, as molecular methods may detect difficult-to-culture bacteria but may in turn miss potentially pathogenic bacteria in low abundance, as these may not significantly alter microbial communities.
There are several limitations to our study. First, based on the ATS/ERS ILD classification into different groups, subjects presented with a certain degree of heterogeneity within this study. Nevertheless, we feel that selecting the five groups, we have compared clinically relevant populations. Second, we did not perform extensive ultra-deep sequencing of viruses and fungi in the collection of samples. Although this has recently been shown to be feasible, fungi identified by this technique were mostly cultivated and known.31 We routinely searched for 15 respiratory viruses, for Pneumocystis jirovecii by immunofluorescence and PCR, measured galactomannan levels and cultured fungi and mycobacteria. We are therefore confident that we did not neglect clinically relevant and currently known pathogens. Third, the number of patients included in this feasibility study is relatively low; therefore, a potential added benefit of characterising microbiota to distinguish between presence of a relevant pathogen and non-infectious clinical worsening in an ILD patient requires further evaluation in prospective, larger studies. Finally, this study includes patients with exacerbated IIP and non-IIP. As we did not include samples from IIP and non-IIP patients during disease stability, we were therefore not able to investigate longitudinal microbiota changes during the actual exacerbation within this study. Nevertheless, considering that we did not detect significant microbiota differences in exacerbated ILD patients compared with normal controls, we hypothesise that changes from an ILD-driven microbiota towards a ‘normal’ microbiota during exacerbation is unlikely and that the presence of a ‘normal’ microbiota during times of stability would be more probable. In agreement with this hypothesis, significant changes in community structure or diversity were not observed at the time of exacerbation in adults with cystic fibrosis.32 ,33
We conclude that 16S rRNA gene-based pyrosequencing is able to characterise microbial communities in upper and lower airways in ILD patients, but the diagnosis of IIP, non-IIP, sarcoidosis or during PCP per se did not alter the overall lung microbiota. Analysis of the topographical continuity along the respiratory tract may provide important additional information to assist the physician in understanding the underlying cause of clinical worsening in ILD patients, a process that will always involve distinguishing infectious from non-infectious causes of inflammation. A potential future application of 16S rRNA gene-based pyrosequencing is the search of hitherto insufficiently known bacteria that may be relevant in the pathogenesis and course of pulmonary disease.
Acknowledgments
We thank the Professors Jean-Francois Dufour, Martin Fey, Felix Frey, Thomas Geiser, Paul Mohacsi and Peter Villiger for their help in providing clinical data and Cornelia Wyss, Monika Wüthrich and Liselotte McEvoy for their assistance with patient recruitment and administrative tasks. We are also grateful to Irène Stutz and Heidi Pfäffli for technical assistance and sample processing.
References
Footnotes
CvG and MH contributed equally.
Kathrin Mühlemann has since died.
-
Contributors Conception and design: CG, SDB, KM, MG-H. Acquisition of data: CG, SDB, WQ, SW, AC, PD, CvG, MG-H. Analysis and interpretation: CG, SDB, CvG, MG-H. Drafting the manuscript: CG, SDB, WQ, MG-H, CvG, MH.
-
Funding This study was supported by unrestricted grants (AstraZeneca, Roche, MSD, Essex Chemie Switzerland, Wyeth, Astellas to C.G.), the Swiss National Science Foundation (M.D.-Ph.D. scholarship 323500-119214 to S.D.B. and Sinergia CRSII3-141875 to M.H. and C.v.G) and the 2012 Award of the Swiss Society of Hospital Hygiene & the Swiss Society for Infectious Diseases (to M.H.).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Kantonale Ethikkommission Bern, approval Nr. 153/08.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves