Article Text

Abstract
Background Patients with allergic asthma have exacerbations which are frequently caused by rhinovirus infection. The antiviral tryptophan-catabolising enzyme indoleamine 2,3-dioxygenase (IDO) is induced by interferon-γ and suppressed by Th2 mediators interleukin (IL)-4 and IL-13. We hypothesised that local IDO activity after viral airway infection is lower in patients with allergic asthma than in healthy controls.
Objective To determine whether IDO activity differs between patients with allergic asthma and healthy individuals before and after rhinovirus infection.
Methods Healthy individuals and patients with allergic asthma were experimentally infected with low-dose (10 TCID50) rhinovirus 16. Blood, bronchoalveolar lavage fluid and exhaled breath condensate (for mass spectrometry by UPLC-MS/MS) were obtained before and after rhinovirus challenge.
Results IDO activity was not induced by rhinovirus infection in either group, despite increases in cold scores. However, baseline pulmonary IDO activity was lower in patients with allergic asthma than in healthy individuals. In contrast, systemic tryptophan and its catabolites were markedly higher in patients with allergic asthma. Moreover, systemic quinolinic acid and tryptophan were associated with eosinophil cationic protein (r=0.43 and r=0.78, respectively) and eosinophils (r=0.38 and r=0.58, respectively) in bronchoalveolar lavage fluid and peak asthma symptom scores after rhinovirus challenge (r=0.53 and r=0.64, respectively).
Conclusions Rhinovirus infection by itself induces no IDO activity, but the reduced pulmonary IDO activity in patients with allergic asthma at baseline may underlie a reduced control of viral infections. Notably, the enhanced systemic catabolism of tryptophan in patients with allergic asthma was strongly related to the outcome of rhinovirus challenge in asthma and may serve as a prognostic factor.
- Asthma Mechanisms
- Viral infection
- Lung Physiology
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is indoleamine 2,3-dioxygenase-mediated tryptophan catabolism different between patients with allergic asthma and healthy individuals and does tryptophan catabolism change upon experimental rhinovirus infection?
What is the bottom line?
-
Tryptophan catabolism is different between healthy individuals and patients with allergic asthma, and is associated with symptom scores and eosinophilic inflammation after rhinovirus exposure.
Why read on?
-
Our study shows that local tryptophan catabolism markedly differs from systemic tryptophan catabolism.
Introduction
Exacerbations represent a major clinical manifestation of asthma and are characterised by episodes of acute worsening of symptoms such as shortness of breath, cough, wheezing and chest tightness in conjunction with airways obstruction.1 Rhinoviruses are the most prominent pathogens to cause exacerbations in patients with asthma.2–4 The underlying mechanisms leading to these lower respiratory tract symptoms are thought to involve both innate and adaptive antiviral immune responses.5 In particular, Th1 (interferon (IFN)γ) and Th2 (interleukin (IL)-4, IL-5 and IL-13) cytokines produced by CD4 cells were shown to be associated with, respectively, peak expiratory flow and chest symptoms after experimental rhinovirus infection in patients with asthma.6
The expression of the tryptophan-catabolising enzyme indoleamine 2,3-dioxygenase (IDO) is induced by IFNγ and inhibited by IL-4 and IL-13.7 ,8 Considering the predominant Th2 cytokine profile in the lungs of individuals with asthma, it can be postulated that patients with allergic asthma fail to induce adequate IDO activity upon viral airway infection. Impaired IDO activity may play a critical role during virus-induced asthma exacerbations, since IDO has been shown to posses both antiviral9–11 and immunosuppressive properties.12 ,13 Several studies underline the immunosuppressive role of IDO in models of allergic airway inflammation. A study by Hayashi et al14 indicated that CpG-induced IDO expression in the lungs reduces airway hyperreactivity in mice with experimental asthma. In line with this, Taher et al15 showed that allergen immunotherapy induces tolerance in mice through an IDO-dependent mechanism. In humans, von Bubnoff et al16 found that asymptomatic allergy was associated with enhanced systemic IDO activity. Whether IDO activity is impaired in the lungs of patients with asthma is unknown.
In the present study we hypothesised that local IDO activity is impaired in patients with allergic asthma after viral airway infection. To that end, we sought to determine: (1) whether IDO-mediated tryptophan catabolism is different between patients with allergic asthma and healthy individuals; and (2) whether IDO-mediated tryptophan catabolism changes upon experimental rhinovirus infection in both healthy individuals and patients with allergic asthma. For this purpose, tryptophan, its product kynurenine and further downstream metabolites anthranilic acid and quinolinic acid were measured in serum and exhaled breath condensate (EBC) before and after experimental rhinovirus infection in healthy individuals and patients with mild allergic asthma.
Methods
Subjects
Healthy individuals met the following criteria: baseline forced expiratory volume in 1 s (FEV1) >80% predicted, airway responsiveness to methacholine (provocative concentration causing 20% fall in FEV1, PC20) >16 mg/mL, skin prick test negative for 12 common aeroallergens. Patients with mild allergic asthma met the following criteria: a history of episodic chest symptoms, baseline FEV1 >80% predicted, PC20 <8 mg/mL, skin prick test positive for at least one of 12 common aeroallergens. All volunteers were aged between 18 and 40 years, were non-smoking or had stopped smoking >12 months ago with ≤5 pack years, were negative for serum neutralising antibodies against rhinovirus 16 and did not have concomitant disease or (chronic) inflammatory condition that would interfere with this study according to the judgement of a pulmonary physician. Volunteers with an underlying viral airway infection on day −1, as determined by PCR on nasal swabs, were withdrawn from the study. Patients with allergic asthma were not allowed to use asthma medication other than short-acting β2 agonists within 2 weeks prior to the start of the study until day 6 after rhinovirus infection.
Study design
Healthy individuals and patients with stable allergic asthma were included in a prospective study with a parallel-group design (figure 1). Between March 2009 and June 2011, 14 healthy individuals and 14 patients with allergic asthma were experimentally infected with low-dose (10 TCID50) rhinovirus 16 (RV16) as described elsewhere.17 In brief, snap-frozen RV16 was rapidly defrosted and taken up in a total volume of 750 μL sterile saline and directly sprayed into one of the nostrils by a DeVilbiss atomiser (DeVilbiss 286; DeVilbiss, Somerset, Pennsylvania, USA). All volunteers were requested to report common cold (sneezing, nasal discharge, stuffy nose, sore throat, cough, chest pain, fever, chills and headache) and asthma symptoms (breathlessness, wheezing, cough, chest tightness and increase in asthma symptoms during the night) daily.18 Individual symptoms were scored on the basis of severity (0=absent, 1=mild, 2=moderate, 3=severe). In addition, volunteers were also asked to report FEV1 as measured twice daily by a portable spirometer (Piko-1, Ferraris Respiratory Europe, UK) and use of short-acting β2 agonists. Diaries and portable spirometers were collected at the final visit (between days 42 and 56).

Study design. Screening was performed 14–56 days before experimental rhinovirus infection. Venous blood, bronchoalveolar lavage fluid and exhaled breath condensate (EBC) were collected at the indicated time points before and after rhinovirus challenge. Blood to determine rhinovirus neutralisation was collected between days 42 and 56 after exposure to rhinovirus type 16 (RV16).
Sample collections
One day before and 6 days after exposure to RV16, nasal swabs and venous blood were collected from all volunteers. In addition, bronchoalveolar lavage (BAL) fluid and bronchial brushes were collected by a standardised bronchoscopic procedure.19
Rhinovirus PCR and neutralisation assay
Nasal swabs, bronchial brushes and 300 μL aliquots of BAL fluid and resuspended BAL cells were processed for rhinovirus detection by quantitative PCR as described elsewhere.20 For RV16 neutralisation, HEL cells were infected with 100 TCID50 RV16 and cultured in the presence of serially diluted serum obtained at the screening visit or between days 42 and day 56 after RV16 exposure. The presence of cytopathic effect in cell cultures was determined 10 days later. The Reed–Muench method was used to estimate neutralisation titre.21 Volunteers who were PCR-negative for rhinovirus on day 6 and did not show increased serum neutralisation after the final visit were excluded from the analysis.
Analyses of inflammatory parameters
IL-4, IL-5, IL-8, IL-13 and IFNγ were determined by Luminex according to the manufacturer's protocol (Bio-Rad Laboratories, Veenendaal, The Netherlands). Eosinophil cationic protein (ECP) and myeloperoxidase (MPO) were measured by ELISA as described previously.22 Cells in BAL fluid were counted and differentiated using cytospin preparations. Fractional exhaled nitric oxide (FeNO) was measured by a chemiluminescence analyser (Niox Aerocrine AB, Solna, Sweden).23
Exhaled breath condensate (EBC)
EBC was collected 1 day before and 4 days after rhinovirus infection using the ECOScreen 1 (Erich Jaeger, Hoechberg, Germany) following the European Respiratory Society Methodological Recommendations Task Force.24 The EBC was stored at −80°C.
Measurements
Analyses of serum and EBC were performed by ultraperformance liquid chromatography hyphenated to tandem mass spectrometry (UPLC-MS/MS). In addition to tryptophan and metabolites of the kynurenine pathway, we also included arginine and related metabolites in our analysis since Lara et al25 reported differences in arginine metabolome between healthy individuals and those with asthma. A mix of stable isotopically-labelled internal standards was added to 0.5 mL EBC. The sample was dried and reconstituted in 100 µL of eluent and 20 µL was injected in the UPLC-MS/MS. The UPLC-MS/MS is an Acquity-Quattro Premier XE system (Waters, Milford, Massachusetts, USA) operated in the positive ESI mode using MRMs for the preselected analytes and an overall run time of 12 min.
Statistical analysis
The sample size was estimated based on cytokine production in previous studies with RV16,26 showing variation of 20% after RV16 infection in patients with allergic asthma. Fourteen individuals per group were needed to detect an effect of 25% with two-sided significance of p=0.05 and a power of 90%.
Demographic data were compared by the Mann–Whitney U test or Fisher exact test where appropriate. Variables that were measured on a daily basis were analysed by two-way ANOVA with Bonferroni post hoc test. Differences between time points were analysed by the Wilcoxon matched pairs test and differences between patient groups were analysed by the Mann–Whitney U test. For tryptophan and related metabolites we used a Bonferroni-adjusted p value, where tryptophan was considered to be an independent variable and kynurenine, anthranilic acid and quinolinic acid were dependent variables. Serum and EBC were considered as independent compartments. Due to possible disparity between the site of infection and the site of sampling, we performed post hoc analysis for tryptophan metabolism in subjects with PCR-confirmed lower respiratory tract infection. In addition, we performed post hoc analysis for markers of arginine metabolism. All other parameters were predefined as primary or secondary endpoints. Spearman correlation was used to determine associations between amino acid levels, symptom scores and inflammatory markers; p<0.05 was considered significant.
Results
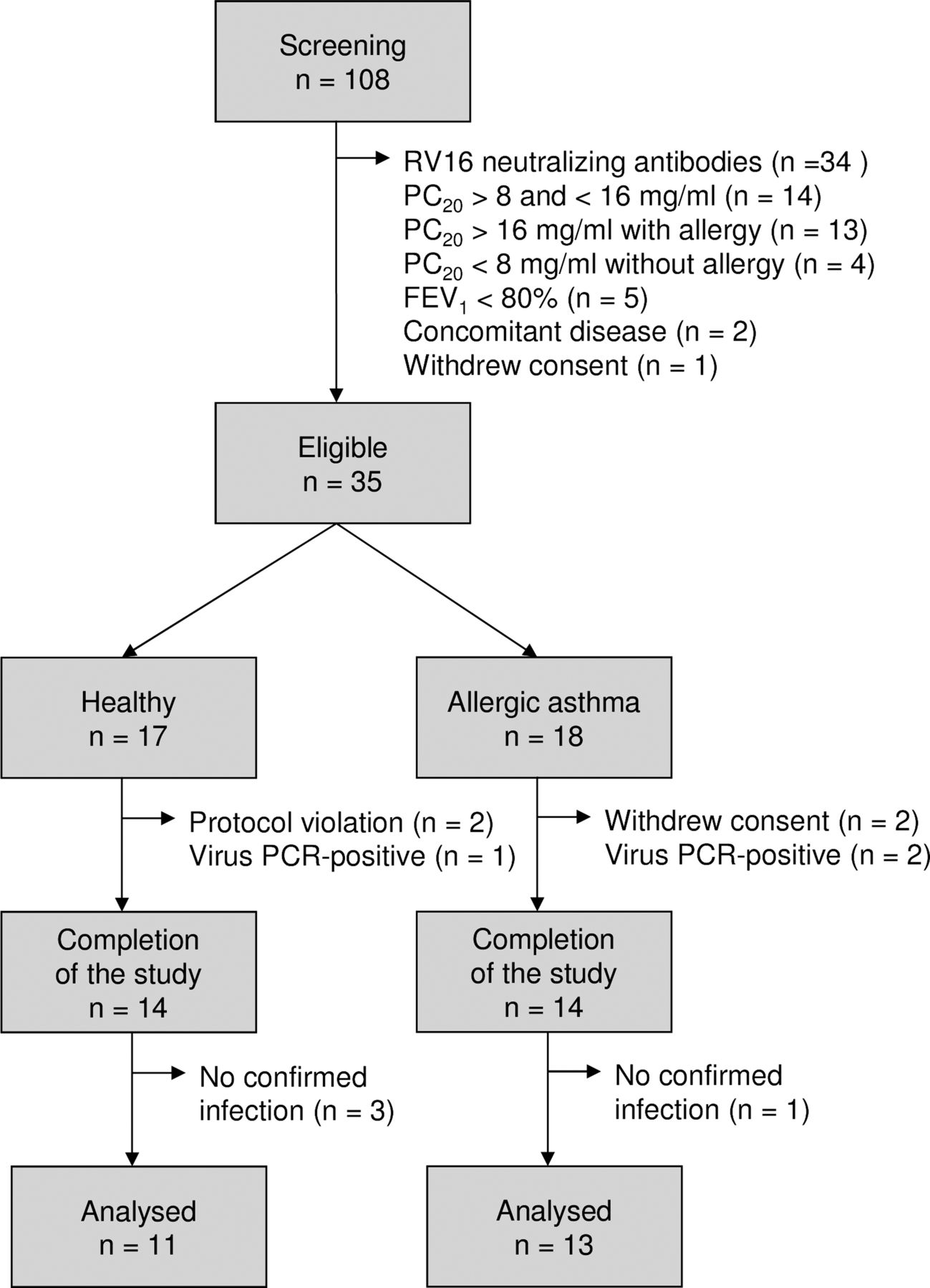
After obtaining informed consent, 108 volunteers were screened, of whom 73 were not eligible for the study because of pre-existing RV16 antibodies or not fulfilling criteria for asthma (figure 2). Thirty-five subjects (17 healthy individuals and 18 with asthma) were enrolled. Three healthy individuals and four patients with allergic asthma were withdrawn from the study after the first bronchoscopy. The baseline characteristics are shown in table 1.
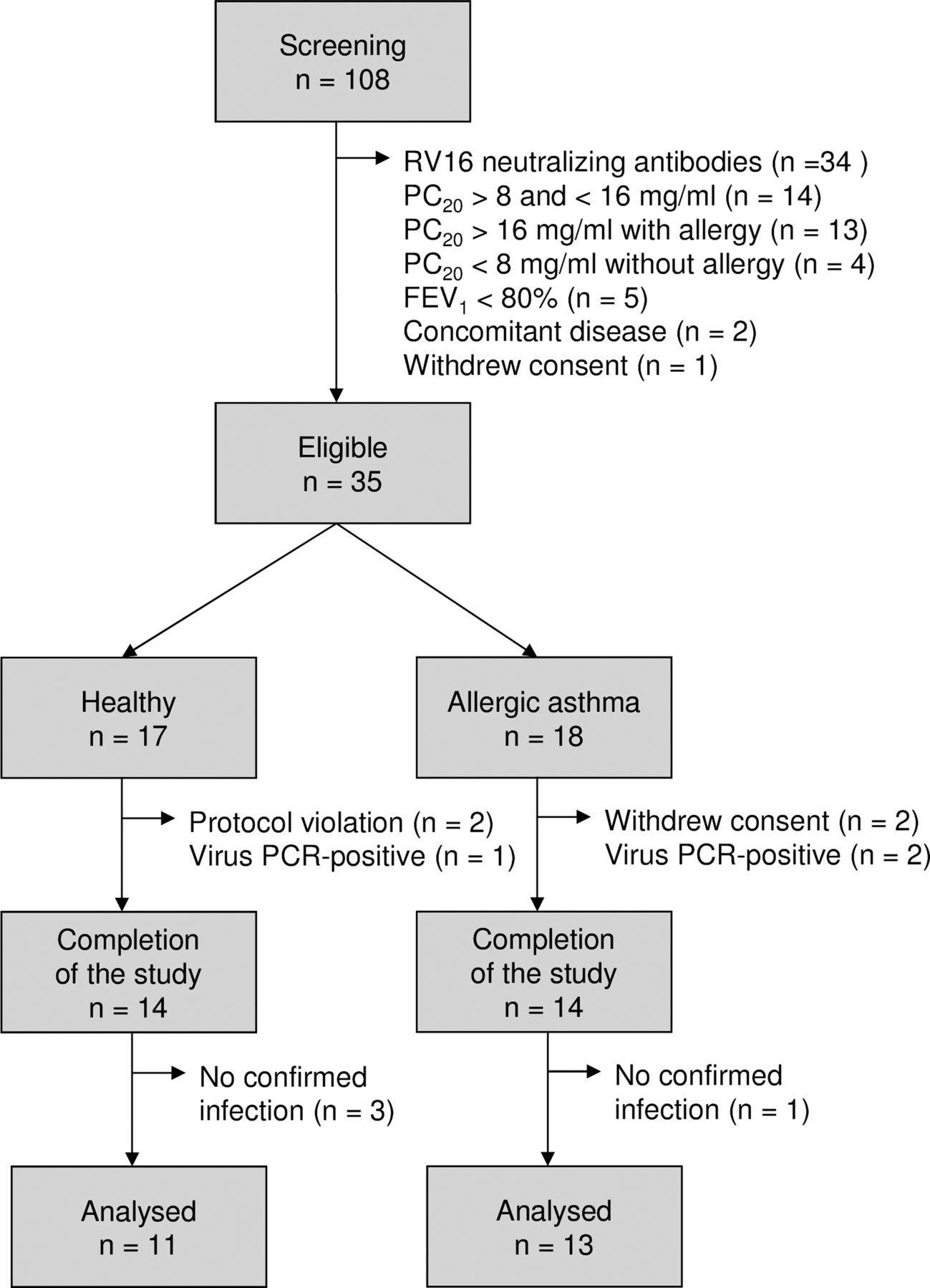
Flowchart for inclusion of healthy individuals and patients with allergic asthma. RV16, rhinovirus type 16; PC20, airway responsiveness to methacholine bromide (provocative concentration causing a 20% fall in FEV1); FEV1, forced expiratory volume in 1 s.
Demographic data of study subjects
Virology
On day 6, nine healthy individuals (64%) and 11 patients with allergic asthma (79%) had a PCR-confirmed infection. The virus load did not differ significantly between the groups (figure 3A). Nine healthy individuals (64%) and 11 patients with allergic asthma (79%) had RV16-neutralising antibodies in the serum collected at the final visit (table 1). Three healthy individuals and one patient with allergic asthma were excluded (figure 2).

Virology and symptoms after experimental rhinovirus infection. (A) Viral RNA copies were determined in upper respiratory tract (nasal swabs) and lower respiratory tract (bronchial brushes, bronchoalveolar lavage (BAL) fluid (without cells) and pelleted BAL cells). (B) Common cold scores, (C) asthma symptom scores and (D) changes in forced expiratory volume in 1 s (FEV1) were reported daily. All data are mean±SE. *p<0.05 and **p<0.01 versus healthy individuals.
Symptoms
Both healthy individuals and those with asthma showed a significant increase in common cold score after experimental infection with a low dose of rhinovirus (figure 3B), with the maximum increase on day 6 (ANOVA: p<0.05 for both groups). Although common cold symptoms appeared higher for patients with allergic asthma at baseline, common cold scores did not differ significantly between healthy individuals and patients with allergic asthma throughout the course of infection.
Experimental rhinovirus infection in patients with allergic asthma resulted in an increase in asthma symptoms (figure 3C) which peaked between days 6 and 8 after exposure (p<0.05 vs healthy individuals). Patients with allergic asthma also showed a significant fall in FEV1 (figure 3D).
Inflammation
ECP levels in BAL fluid were significantly increased after rhinovirus infection in patients with allergic asthma but not in healthy individuals (table 2). FeNO, ECP and eosinophils in BAL fluid were significantly higher in patients with allergic asthma than in healthy individuals at day 1 before and day 6 after rhinovirus exposure (table 2). IL-8, MPO and BAL fluid cells other than eosinophils were not significantly different between the two time points and between the two groups (table 2). IL-4, IL-5, IL-13 and IFNγ were undetectable in BAL fluid.
Inflammatory parameters in bronchoalveolar lavage fluid
Local and systemic amino acid metabolism
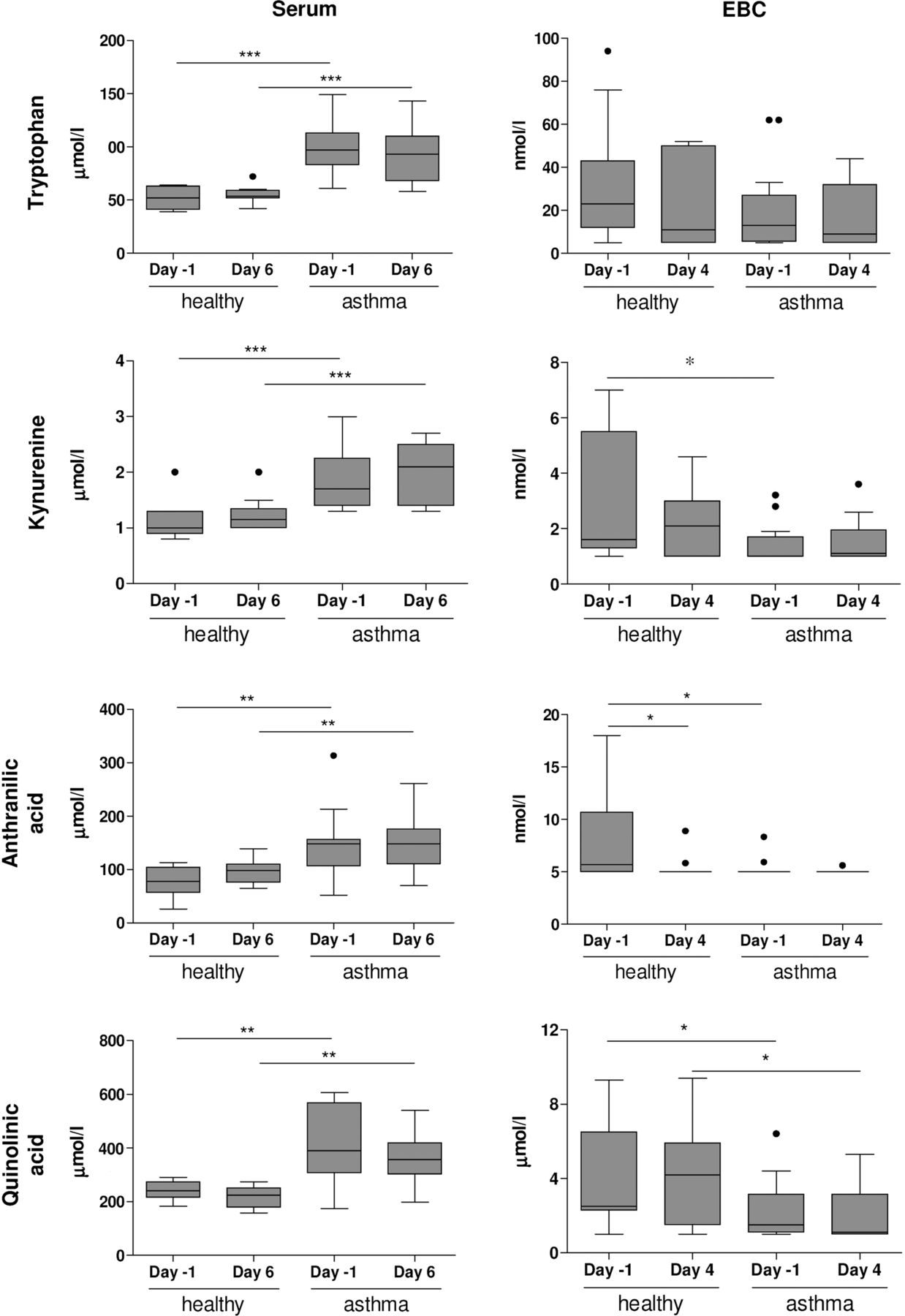
Patients with allergic asthma showed significantly higher levels of tryptophan, kynurenine, quinolinic acid and anthranilic acid in serum than healthy individuals (figure 4) and were not affected by exposure to RV16. In contrast, patients with allergic asthma showed lower levels of kynurenine (day −1), anthranilic acid (day −1) and quinolinic acid (day 4) in EBC than healthy individuals (figure 4). Tryptophan levels in EBC were not different between healthy individuals and patients with allergic asthma. Similar differences were observed when subjects without PCR-confirmed lower respiratory tract infections were excluded (see online supplementary figure S1).
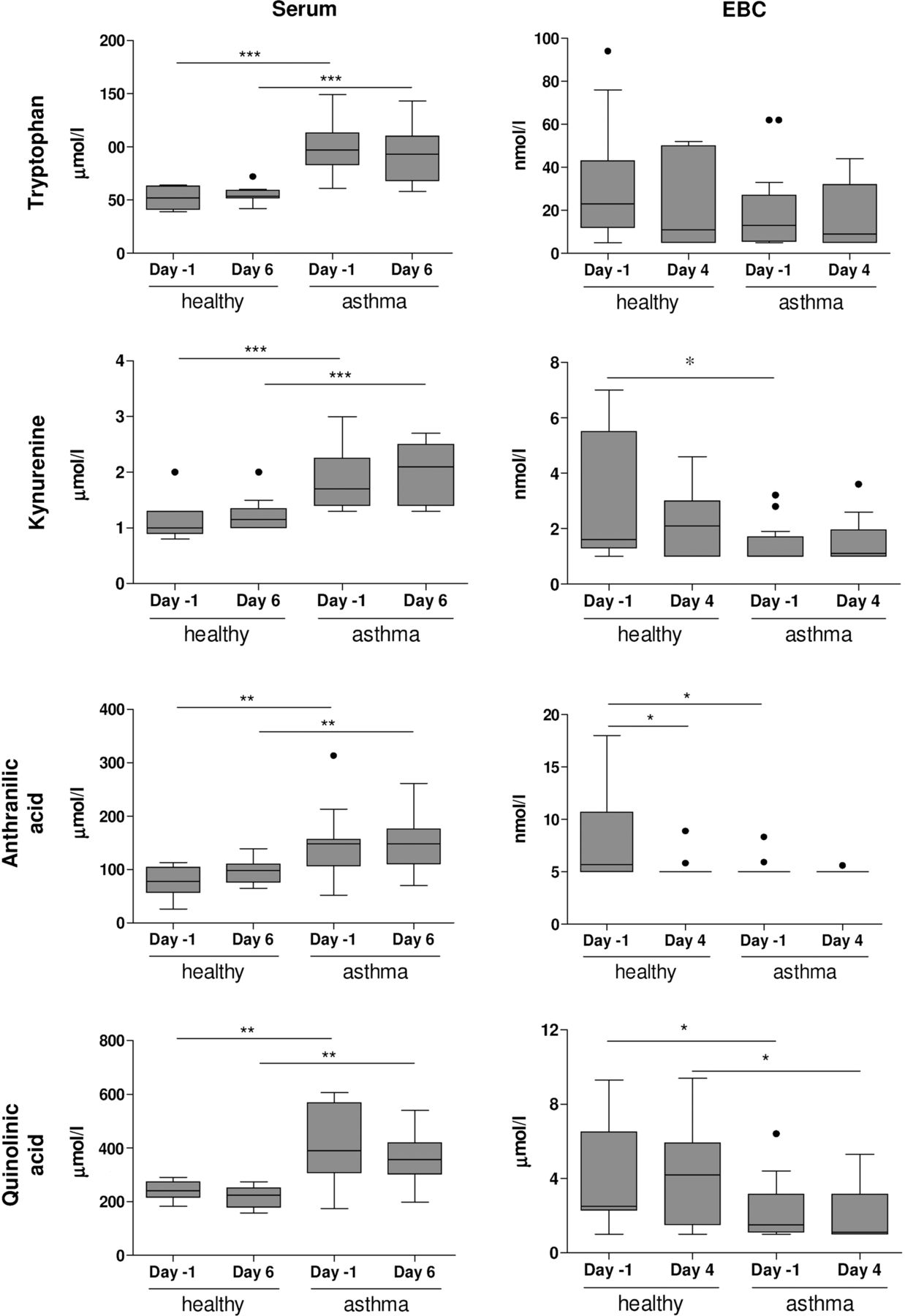
Tryptophan metabolome in exhaled breath condensate (EBC) and serum. Tryptophan, kynurenine, anthranilic acid and quinolinic acid were determined in serum (day −1 and day 6) and EBC (day −1 and day 4) of healthy individuals and patients with allergic asthma. Concentrations are shown as nmol/L or μmol/L (mean±SE). *p<0.05, **p<0.01 and ***p<0.001 versus healthy individuals (Bonferroni-adjusted p values). Kynurenine (day −1, p=0.0186), anthranilic acid (day −1, p=0.0197) and quinolonic acid (day 4, p=0.0166) were lower in EBC from patients with allergic asthma than in EBC from healthy individuals (unadjusted p values). Detection limits are: tryptophan (5 nmol/L), kynurenine (1 nmol/L), quinolinic acid (1 nmol/L) and anthranilic acid (5 nmol/L).
Post hoc analysis revealed significantly higher serum arginine levels for patients with allergic asthma than for healthy individuals, which was not affected by rhinovirus infection (see online supplementary figure S2). Patients with allergic asthma had lower levels of arginine and markedly higher levels of urea in EBC than healthy individuals, while lower ornithine levels in EBC were lower after rhinovirus infection (p<0.05 vs healthy individuals).
Tryptophan catabolism and rhinovirus-induced exacerbation
Quinolinic acid was significantly lower in EBC from patients with allergic asthma than healthy individuals after rhinovirus challenge. However, quinolinic acid levels in EBC did not show an association with virus load, asthma symptom scores or eosinophilic inflammation in the lung. Systemic levels of quinolinic acid were associated with peak asthma symptom scores (Spearman r=0.5298, p=0.0078) and ECP in BAL fluid (r=0.4321, p=0.0395). The associations of systemic tryptophan with peak asthma symptom scores (r=0.6421, p=0.0007), percentage BAL fluid eosinophils (r=0.5840, p=0.0027) and ECP in BAL fluid (r=0.7824, p<0.0001) were even stronger (figure 5). Similar correlations were observed when subjects without PCR-confirmed lower respiratory tract infections were excluded (see online supplementary figure S3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association between tryptophan metabolism and eosinophilic inflammation. Analyses of the correlation between systemic quinolinic acid (left) and systemic tryptophan (right) with (A) percentage eosinophils in bronchoalveolar lavage (BAL) fluid, (B) eosinophil cationic protein concentration in BAL fluid and (C) peak asthma symptom scores after rhinovirus exposure in patients with asthma (solid dots) and healthy individuals (open dots). Spearman correlation coefficients (r) and p values are given in each panel.
Discussion
In the present study we showed that a low-dose rhinovirus infection (10 TCID50) induced characteristics of a mild exacerbation in patients with asthma, reflected by increased asthma symptoms, reduced lung function and enhanced eosinophilic inflammation. Although IDO activity did not change upon rhinovirus infection, patients with allergic asthma already showed lower IDO activity in the lungs at baseline than healthy individuals. Conversely, enhanced levels of cytotoxic metabolites and tryptophan were found in the serum of patients with asthma which were associated with eosinophilic airway inflammation and peak asthma symptom scores after rhinovirus infection. There was no association between tryptophan catabolism in the airways and exacerbation parameters.
Tryptophan is catabolised via multiple pathways. Tryptophan 2,3-dioxgenase (TDO), IDO1 and IDO2 are known to enhance the degradation of tryptophan into N-formyl-kynurenine, which is rapidly converted into kynurenine27 ,28 and further degraded into 3-hydroxy-kynurenine, anthranilic acid, 3-hydroxy-anthranilic acid and quinolinic acid. TDO is constitutively and specifically expressed in the liver and maintains circulating tryptophan levels within a physiological range.29 In contrast, IDO1 and IDO2 are hardly expressed under normal conditions but are induced by inflammatory mediators.8–11 ,29 We and others have previously shown that viral airway infection leads to pulmonary expression of IDO.30–32
The lower levels of kynurenine and quinolinic acid in EBC indicate that pulmonary IDO activity is impaired in patients with allergic asthma. This reduced IDO activity in patients with allergic asthma may relate to reduced IDO expression but also to nitric oxide, which is known to bind the heme group of IDO33 thereby limiting its enzymatic activity. Of note, patients with allergic asthma have higher fractions of exhaled nitric oxide.
In contrast to our expectation, rhinovirus infection did not enhance IDO activity in either healthy individuals or those with asthma. This observation may be explained in several mutually non-exclusive ways. First, the observation that kynurenine and quinolinic acid are present in EBC indicates that IDO is already expressed in the pulmonary compartment under normal conditions, which may have masked a modest effect induced by rhinovirus infection. Second, the volunteers were challenged with a low dose of RV16. Despite increased common cold and asthma symptoms, the inflammatory response may have been insufficient to induce IDO expression. RV16 showed an increase in ECP and IL-8 in BAL fluid of individuals with asthma, but mediators that have been described to modulate IDO expression were not detectable. It should be noted that low-dose rhinovirus exposure resulted in similar common cold and asthma symptom scores as those described for high-dose rhinovirus exposure,5 ,18 ,26 which argues against the low-dose rhinovirus as a possible explanation. In addition, we only included subjects with confirmed viral infection—that is, subjects without neutralising antibodies against RV16—at the start of the study. The fact that animal studies showing pulmonary IDO expression have been performed with influenza30–32 might indicate that the virulence of the virus is more relevant for pulmonary IDO expression. Prospective observational studies are required to determine whether influenza induces IDO in human airways.
In serum, patients with allergic asthma showed significantly higher levels of tryptophan and cytotoxic metabolites than healthy individuals, which confirms previous observations by Warraki et al.34 Interestingly, we observed a significant association between quinolinic acid and percentage eosinophils and ECP in BAL fluid. These findings indicate that quinolinic acid may be used as a marker for local eosinophilic inflammation in response to rhinovirus infection. The higher tryptophan levels in serum from patients with allergic asthma is in line with a higher OR of having asthma with high tryptophan levels in plasma as reported by Fogarty et al.35 It should be noted that this study only included patients who received daily inhaled steroids whereas our study only included patients without inhaled or oral corticosteroids. Whether the enhanced systemic levels of kynurenine catabolites in patients with asthma have physiological consequences such as reduced T cell responses remains to be determined.
For markers of arginine metabolism we obtained a similar systemic increase as previously shown by Lara et al.25 In line with our findings for quinolinic acid, we observed lower arginine levels in EBC from patients with asthma. Healthy individuals but not those with asthma showed increased ornithine levels in EBC upon rhinovirus challenge, which may point towards a role for arginase and/or alternatively activated macrophages in host defence towards viral airway infection in healthy individuals. Together these findings indicate that the amino acid catabolism in the systemic and pulmonary compartments can be quite distinct.
In the present study we exposed volunteers with a 1000-fold lower dose than in previous studies on rhinovirus-induced asthma exacerbations.6 ,18 ,26 Although common cold and asthma symptoms peaked 2 days later, the symptoms scores were similar to those described for the high viral inoculum. Twenty-four individuals (86%) had a confirmed viral infection as reflected by detection of virus by quantitative PCR on day 6 or RV16 neutralisation between days 42 and 56 after viral infection. These data indicate that low-dose RV16, which is more likely to reflect a natural common cold than high-dose RV16, can be used to provoke common cold and asthma symptoms in patients with mild allergic asthma. The fact that we did not observe differences in viral load between healthy individuals and those with asthma must be interpreted with caution since viral load was determined at a single time point.
The design of this study has some important limitations. First, the male/female ratio was different for healthy individuals (1/13) and patients with asthma (7/7), which might have an impact on the results. However, there were no differences between men and women with asthma with respect to systemic amino acid levels. In addition, the differences between healthy individuals and those with asthma were still significant when only female volunteers were included in the statistical analysis. We therefore conclude that the male/female ratio is not a substantial confounding factor.
Second, the healthy individuals and those with asthma differed in bronchial hyperresponsiveness to methacholine and also in allergy status. We specifically chose to select for allergies in the asthma group since we hypothesised that the balance between IFNγ and Th2 cytokines would affect the expression of IDO and, hence, also tryptophan catabolism. Since these cytokines were undetectable or hardly detectable in BAL fluid, we were unable to observe an association between cytokine responses and tryptophan turnover. It is therefore not possible to conclude whether the differences between healthy individuals and patients with allergic asthma are due to allergy, asthma or a combination of both. Further research is required to identify the contribution of the allergic status.
Third, volunteers underwent bronchoscopy 1 day before exposure to RV16. This procedure probably affected common cold scores and asthma symptom scores, particularly for individuals with allergic asthma. Nonetheless, both healthy individuals and patients with allergic asthma showed a significant increase in common cold symptoms and, in addition, patients with asthma showed a significant increase in asthma symptoms. These findings indicate that rhinovirus infection was successful in these volunteers. It is important to note that blood and EBC samples were always collected before the bronchoscopic procedure. The second EBC sample was collected on day 4 and the second blood sample on day 6. Differences between EBC and plasma are therefore only mentioned if these differences were already present on day 1 prior to infection when EBC and blood were collected in parallel.
In conclusion, the present study shows that patients with mild allergic asthma have lower IDO activity in the pulmonary compartment than controls, which is not enhanced by experimental rhinovirus infection. Systemic levels of tryptophan and downstream metabolites of TDO/IDO-mediated tryptophan degradation were markedly higher in patients with allergic asthma and were associated with eosinophilic inflammation and asthma symptom scores during experimental rhinovirus infection. The underlying cause of these newly observed differences in tryptophan metabolism remains to be elucidated.
Acknowledgments
We would like to thank Katrien Grünberg (VU Medical Center, Amsterdam, the Netherlands), José de Kluijver (Leiden University Medical Center, Leiden, the Netherlands) and Joseph Footitt (Imperial College, London, UK) for advice on the rhinovirus infection protocol and the members of the RESOLVE research team: Marieke Berger, Niki Fens, David Yick, Ariane H Wagener, Marijke Amelink, Herre J Reesink, Paul Bresser, Peter W A Kunst, Lisette N Venekamp, Christof J Majoor, Reindert P van Steenwijk, René E Jonkers. We would also like to thank Dr A H Zwinderman for advice on statistical analysis of the data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Collaborators Marieke Berger; Niki Fens; David Yick; Ariane H Wagener; Marijke Amelink; Herre J Reesink; Paul Bresser; Peter W A Kunst; Lisette N Venekamp; Christof J Majoor; Reindert P van Steenwijk; René E Jonkers.
-
Contributors KFvdS was involved in the conception and design of the study, acquisition, analysis and interpretation of the data and wrote the manuscript. MAvdP, WK, AD, HWvE, JAK, RM and KCW were involved in acquisition, analysis and interpretation of the data. SLJ, JSvdZ, PJS and RL were involved in the conception and design of the study and interpretation of the data. All authors have seen and approved the content of the manuscript prior to submission. The RESOLVE research team members were actively involved in clinical aspects related to the conduct of this study.
-
Funding This study was financially supported by a grant from the Netherlands Asthma Foundation (grant no 3-2-07-012). SLJ is supported by ERC FP7 advanced grant 233015, a Chair from Asthma UK (CH11SJ) and MRC Centre grant G1000758.
-
Competing interests None.
-
Ethics approval The study was approved by the Medical Ethics Committee of the Academic Medical Center in Amsterdam, The Netherlands and registered at the Netherlands Trial Registration (no.1677).
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.