Article Text

Abstract
Background Respiratory syncytial virus (RSV) remains the leading cause of serious viral bronchiolitis and pneumonia in infants and young children throughout the world. The burden of disease is significant, with 70% of all infants being infected with RSV within the first year of their life. 40% of those children discharged from hospital have recurrent, repeated respiratory symptoms and wheezing for at least 10 years. The infection is also an important illness in the elderly and immunocompromised individuals. Ongoing symptoms relate to continued lung inflammation. One cytokine that is associated with RSV infection is IL-1β, but the mechanism of activation remain unclear.
Objectives In the current study, we set out to decipher the molecular mechanisms of RSV-induced inflammasome activation.
Methods and results Using deletion mutants of the virus and measuring IL-1β secretion, as well as caspase 1 expression via western blotting, we demonstrate that the NLRP3 inflammasome is activated through the small hydrophobic (SH) RSV viroporin which induces membrane permeability to ions or small molecules. Confocal microscopy revealed that during virus infection, SH seems to accumulate within lipid rafts in the Golgi compartments.
Conclusions Upon RSV infection, SH gets localised in the cell membranes and intracellular organelle membranes, and then induces permeability by disrupting membrane architecture, thus leading us to believe that formation of viral ion channels in lipid bilayers of cells is a viral recognition pathway used by the host to signal inflammasome activation.
- Airway Epithelium
- Innate Immunity
- Respiratory Infection
- Viral infection
Statistics from Altmetric.com
Key messages
What is the key question?
-
How does respiratory syncytial virus (RSV) trigger inflammasome activation?
What is the bottom line?
-
We feel that the findings in this manuscript are highly novel. Our data reveals that upon RSV infection, the RSV small hydrophobic protein accumulates in the Golgi within lipid raft structures, forming ion channels, which trigger the translocation of NLRP3 from the cytoplasm to the Golgi.
Why read on?
-
We can conclude that these intracellular raft structures are crucial for sensing cellular stress imposed by imbalances in ionic concentrations, and could be a general viral recognition pathway that can be utilised by the infected host cell to signal the activation of the NLR inflammasomes.
Introduction
Respiratory syncytial virus (RSV) remains the leading cause of serious viral bronchiolitis and pneumonia in infants and young children throughout the world. The burden of disease is significant, with 70% of all infants being infected with RSV within the first year of their life.1 It is the most common cause of hospitalisation in infants, and of acute respiratory failure in paediatric intensive care units. Forty per cent of those children discharged from hospital have recurrent, repeated respiratory symptoms and wheezing for at least 10 years.2 The infection is also important in the elderly and immunocompromised individuals. The pathology of the virus is associated with innate immunity and skewed immune responses towards a Th2 phenotype.3 ,4
One cytokine that is associated with RSV infection is IL-1β. Although this cytokine has been shown to be secreted by RSV-infected airway cells,5 the question that remains is in respect of the mechanism of inflammasome activation. It is now emerging that inflammasomes are multiprotein complexes that act as a platform for the activation of caspase 1 which, in turn, cleaves pro-IL1β and pro-IL18 resulting in their secretion.6
Since inflammasomes have been shown to be triggered by diverse ligands, it has been suggested that two or potentially more signals are required for full activation. The first, or priming signal, can by triggered from a transciptionally active PRR or cytokine receptor, this leads to transcriptional activation of the genes encoding pro-IL1β and pro-IL18.7 The second signal is triggered in response to various stress signals associated with damaged self.6
The question that arises is how is IL-1β triggered in response to RSV infection, and which is the ‘priming’ step that leads to IL-1β secretion. A very recent study by Segovia et al8 has shown that RSV triggers NLRP3 activation via reactive oxygen species (ROS) production. Although this study has provided some clues, it has not revealed which part of the virus triggers this inflammation, and the mechanism of NLRP3 activation remains unknown.
In the current study, we have attempted to shed more light into the mechanisms of RSV-induced inflammasome activation. Our investigations have revealed that the RSV small hydrophobic (SH) viroporin which induces membrane permeability to ions or small molecules9 is essential in triggering the NLRP3 inflammasome in RSV infections.
In our study, pharmacological treatment of RSV-infected cells with drugs that inhibit viral ion channels and lipid raft disruptors, blocked inflammasome activation showing that lipid raft structure in intracellular compartments plays an important role in inflammasome activation. Our study demonstrates that upon RSV infection, the RSV SH viroporin accumulates in the Golgi within lipid raft structures, possibly forming ion channels, which trigger the translocation of NLRP3 from the cytoplasm to the Golgi.
Methods
Viruses
Human respiratory syncytial virus strain A2, human herpes simplex virus 1 strain MacIntyre, encephalomyocarditis virus (tissue culture adapted), vesicular stomatitis Orsay virus were originally purchased from American Tissue Culture Collection. The viruses were propagated and titrated in African green monkey cells. The mutant virus rgRSV-GF (ΔSH RSV) was provided by Drs Mark Peeples and Peter Collins.10 The ΔSH RSV mutant has been shown in previous studies to grow and replicate efficiently in cell culture (Bukreyev et al, J Virol 1997). We have also performed growth curves of this mutant, and we see that it is replication and growth competent similar to the wild type. These viruses were stored at −80°C.
Cells
Primary human lung epithelial cells were obtained from an adult male (TCS cell works, UK).They were passaged and maintained in the epithelial cell medium provided by TCS.
Chemicals/reagents
All fine chemicals were obtained from Sigma (UK). TLR2, TLR4 specific mAbs, were obtained from Hycult (Denmark). TLR3, TLR9, TLR7 and TLR8 specific antibodies were purchased from Santa Cruz Biotechnology (California, USA). RIG-I specific antibodies were purchased from Abcam (Cambridge, UK). Rabbit polyclonal caspase 1 p10 antibody, rabbit polyclonal NLRP3 antibody and apoptosis-associated speck-like protein (ASC) goat polyclonal antibody were purchased from Santa Cruz Biotechnology (California, USA). IL1β antibody (NBP1-42767) was obtained from Novus (USA). Rabbit polyclonal to IL1β was purchased by abcam, (USA). Polyclonal antibody (AbSH), specific for the SH protein of the RSV,11 was a kind gift from Professor Biao He.
RNA interference
RNA interference was used in order to silence the RIG-I, TLR7, TLR3, TLR4, TLR8 and TLR2 genes. Different psiRNA clones were generated using the psiRNA-h7SK vector from Invitrogen; the most efficient was against the sequence: for RIG-I, GGAAGAGGTGCAGTATATT, for TLR7 GGGTATCAGCGTCTAATATCA, for TLR8 GACCAACTTCGATACCTAAA, for NLRP3 GGAAGTGGACTGCGAGAAGTT, for TLR3, GAGTTAGATATGCGCTTTAAT, for TLR4 GCCAGGAGAACTACGTGTGAA for TLR2 GTCAATTCAGAACGTAAGTCA.
Human lung epithelial cells (1×105) were seeded in six-well plates and transfected with 0.5 μg of psiRNA for either RIG-I, or TLR3, TLR7, TLR4, TLR8 or scrambled siRNA as a control using Lipofectamine 2000 (Invitrogen). After 48 h, the level of silencing was determined by western blotting.
Confocal microscopy
Human lung epithelial cells on microchamber culture slides (Lab-tek, Gibco), were incubated with RSV multiplicity of infection (MOI 5), and were subsequently rinsed twice in PBS/0.02% Bovine serum albumin (BSA), prior to fixation with 4% formaldehyde for 15 min. The cells were fixed in order to prevent potential reorganisation of the proteins during the course of the experiment. Cells were permeabilised using PBS/0.02% BSA/0.02% saponin and labelled with antibodies for NLRP3, ASC directly labelled with the appropriate fluorophore. Golgi was stained with GM130 mAb, followed by Alexa 488-Fab mouse-specific Ig. Cells were imaged on a Carl Zeiss, Inc LSM510 META confocal microscope (with an Axiovert 200 fluorescent microscope) using a 1.4 NA 63× Zeiss objective. The images were analysed using LSM 2.5 image analysis software (Carl Zeiss, Inc).
Optimisation of drug concentrations to inflammasome
Human lung epithelial cells were seeded onto six-well plates. The following day, cells were infected with RSV at a MOI 5 in medium containing the appropriate concentration of each drug for 60 min (5-N-ethyl-N-isoproamiloride 25 μM, Benzamil 50 μM, Verapamil 50 μM, nystatin 25 μM, amantadine or rimantadine 6.5 μM, hexamethylene amiloride (HMA), 50 μM).
Supernatant containing virus was removed and the cells were washed. Fresh medium containing the drugs was added and the cells were incubated for a total of 12 h. (Viability of cells was determined by using 0.2% trypan blue while examining cells under a microscope).
Cell-free supernatant was collected at 12 h postinfection and analysed for IL-1β by cytometric bead array (CBA). In some cases, supernatants and cell extracts were collected for western blot analysis.
Fluorescence resonance energy transfer (FRET)
FRET is a non-invasive imaging technique used to determine molecular proximity. FRET can occur over 1–10 nm distances, and effectively increases the resolution of light microscopy to the molecular level. It involves non-radiative transfer of energy from the excited state of a donor molecule to an appropriate acceptor. The rate of energy transfer is inversely proportional to the sixth power of the distance, between donor and acceptor. The efficiency of energy transfer (E) is defined with respect to r and R0, the characteristic Forster distance by 1
1
FRET will be measured in terms of dequenching of donor fluorescence after complete photo-bleaching of the acceptor fluorophore. Increased donor fluorescence after complete destruction of the acceptor indicates association between the two molecules of interest.
Results
RSV infection activates the NLRP3 inflammasome
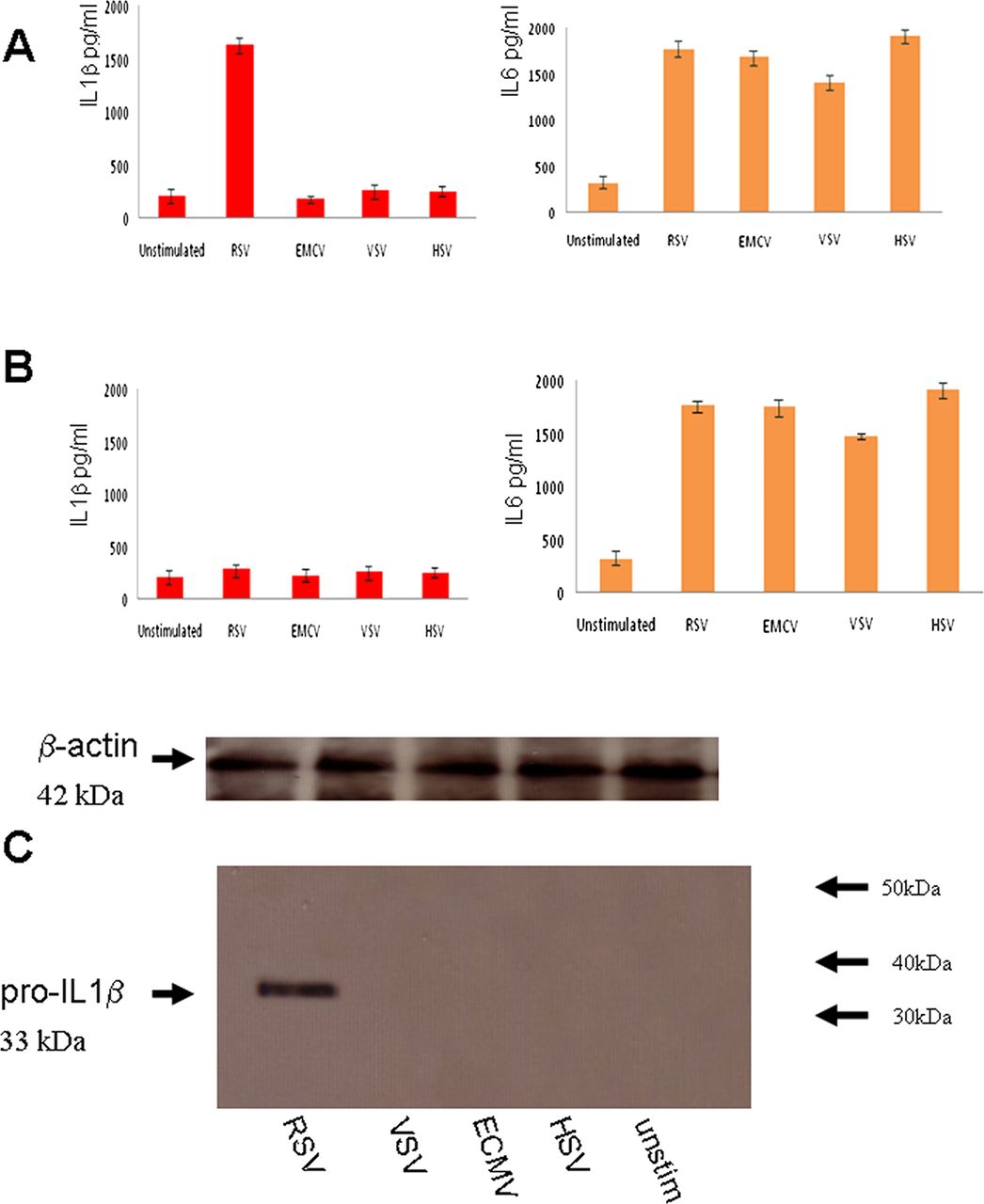
In order to determine if RSV is detected by the inflammasome, we investigated inflammasome activation by measuring IL1β secretion from primary RSV-infected lung epithelial cells. RNA viruses, such as vesicular stomatitis virus (VSV), and encephalomyocarditis virus (ECMV), as well as a DNA virus Herpes simplex virus (HSV1), were also compared with RSV.
The data showed that RSV infection resulted in IL1β secretion within 6 h (figure 1A). IL1β secretion was not observed in infections with the other viruses. To verify if IL1β secretion was NLRP3-specific, NLRP3 expression was knocked down by psiRNA which resulted in a reduction in IL1β secretion (figure 1B), thus verifying that RSV is detected by the NLRP3 inflammasome. IL-6 secretion was measured as a control for the psiRNA effect since it is independent of inflammasome activation but is a known marker for NF-kB activation. All the tested viruses triggered IL-6 secretion, but only RSV was detected by the inflammasome in these unprimed cells.

Respiratory syncytial virus (RSV) infection activates the NLRP3 inflammasome. Lung epithelial cells were infected with 5 multiplicity of infection (moi) of RSV or ECMV, or vesicular stomatitis virus (VSV) or HSV1 viruses. Supernatant was collected at 12 h postinfection and analysed for IL1β and IL6 using the CBA bead array system on a FACSCalibur (Becton Dickinson) (A). NLRP3 expression was knocked down by psiRNA and the cells were again infected with 5moi of RSV or ECMV, or VSV or HSV1 viruses. Supernatant was collected at 12 h postinfection, and analysed for IL1β and IL6 using the CBA system (B) Cells extracts from infected lung epithelial cells were analysed for the presence of pro-IL1β by western blotting and loading controls for β-actin are also depicted (C). The data represent the mean ±SD, of three independent experiments.
RSV triggers pro-IL1β secretion
IL1β secretion requires two signals: transcription of pro-IL1β (signal 1) and processing by caspase 1 (signal 2). To examine whether RSV infection provides the transcriptional signal, we infected cells with RSV and examined the pro-IL1β response via western blotting. RSV infection induced a pro-IL1β response, while infection by the other viruses did not (figure 1C).
TLR signalling is required for inflammasome actvation
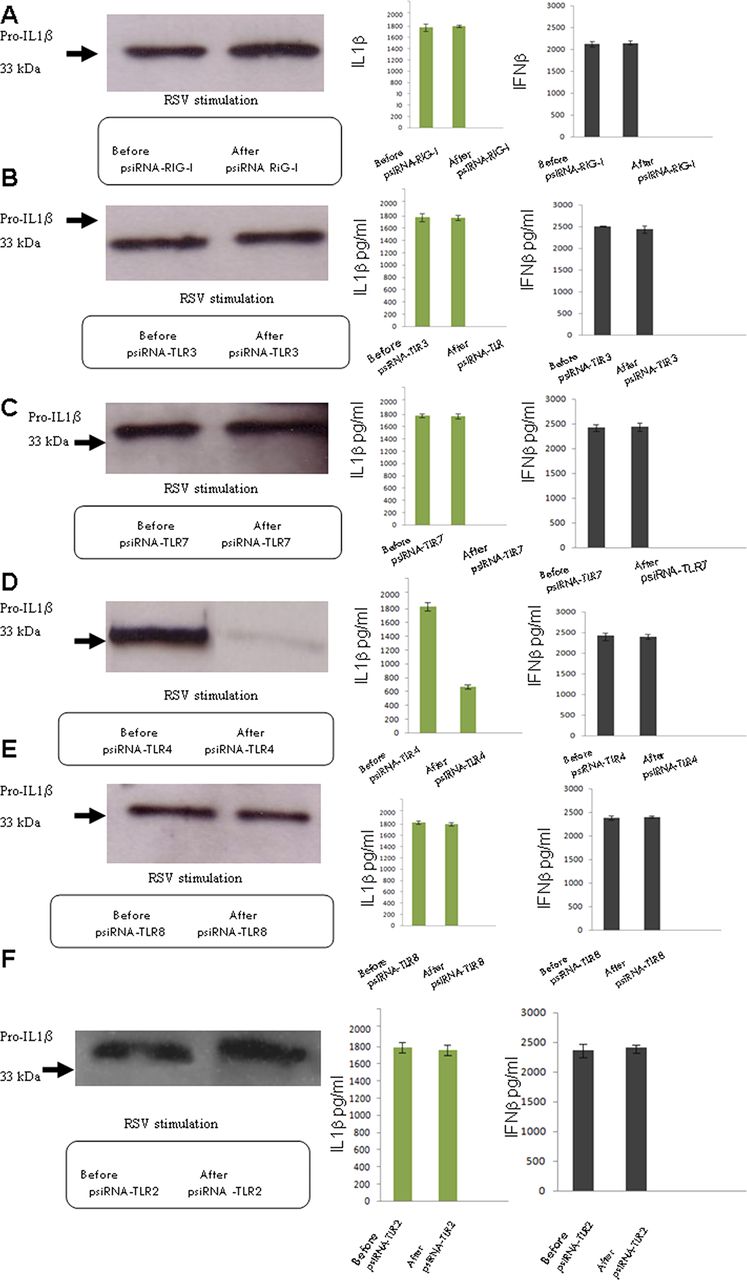
RSV is detected by different PRRs, such as TLR4,12 TLR7,13 TLR3 and RIG-I.14 To determine if they also play a role in inflammasome activation, we individually knocked down RIG-I, TLR3, TLR4, TLR7 or TLR8 in RSV-infected cells as well as TLR2 (figure 2). Only the knocking down of TLR4 resulted in a reduction in expression of pro-IL1β and IL1β. IFNβ secretion was not reduced by knocking down any of these molecules, showing that the IL1β reduction was specific. TLR4, therefore, appears to be required for signal 1, the transcription of pro-ILβ that results in the subsequent release of mature IL1β by RSV infection.

TLR4 provides the first or priming signal of activation. Lung epithelial cells were infected with 5 multiplicity of infection (moi) of respiratory syncytial virus and the presence of pro-IL1β by western blotting was examined, as well as the secretion of IL1β and IFNβ using the CBA system. RIG-I expression was knocked down by psiRNA, and the cells were again infected and examined for pro- IL1β, as well as IL1β and IFNβ secretion (A). TLR3 expression was knocked down by psiRNA and the cells were again infected and tested for pro-IL1β, as well as IL1β and IFNβ secretion (B). TLR7 expression was knocked down by psiRNA and the cells were again infected and tested for pro- IL1β, as well as IL1β and IFNβ secretion (C). TLR4 expression was knocked down by psiRNA and the cells were again infected and tested for pro-IL1β, as well as IL1β and IFNβ secretion (D). TLR8 expression was knocked down by psiRNA and the cells were again infected and tested for pro-IL1β, as well as IL1β and IFNβ secretion (E). TLR2 expression was knocked down by psiRNA and the cells were again infected and tested for pro-IL1β, as well as IL1β and IFNβ secretion (F). The data represent the mean of three independent experiments.
However, some IL1β was still produced leading us to believe that in addition to the established TLR sensors there could yet exist an additional mechanism for triggering IL1β production.
SH-mediated mechanism of inflammasome activation
To discover how RSV infection triggers signal 2, we looked at RSV viral proteins which enhance membrane permeability since It has been shown that the influenza M2 ion channel induces inflammasome activation by modulating the intracellular K+ concentration.15
The RSV small hydrophobic protein has been classified as a viroporin. It alters membrane permeability displaying properties of a cation-selective ion channel.16 ,17 SH is not essential for viral replication in vitro (Bukreyev et al 1997). However, deletion of the SH gene leads to attenuation in mouse and chimpanzee models, suggesting it is important for infectivity in vivo (Bukreyev et al 1997; Whitehead et al 1999).
To test the role of SH in inflammasome activation, an SH deletion mutant RSV rgRSV-GF(ΔSHRSV)10 was used. The ΔSHRSV mutant has been shown in previous studies to grow and replicate efficiently in cell culture.18 The infection with rgRSV-GF failed to induce caspase activation and release of IL1β (figure 3A). However, rgRSV-GF-infected cells released IL6 a non-inflammasome cytokine, IFNβ and pro-IL1β. RSV infection induced caspase 1 production, while rgRSV-GF infection did not. Together, these results indicate that SH plays an essential role in triggering signal 2 leading to caspase 1 production and inflammasome activation (figure 3)

Respiratory syncytial virus (RSV) and ΔSHRSV mediated mechanism of inflammasome activation. Lung epithelial cells were either infected with RSV or ΔSHRSV. Cell supernatant was collected and tested for IL1β (A), IL6 (B) and IFNβ (C) secretion. Cells extracts from infected lung epithelial cells were analysed for the presence of pro-IL1β (D) and Caspase p10 (E) by western blotting. The data presented are the mean±SD of three independent experiments.
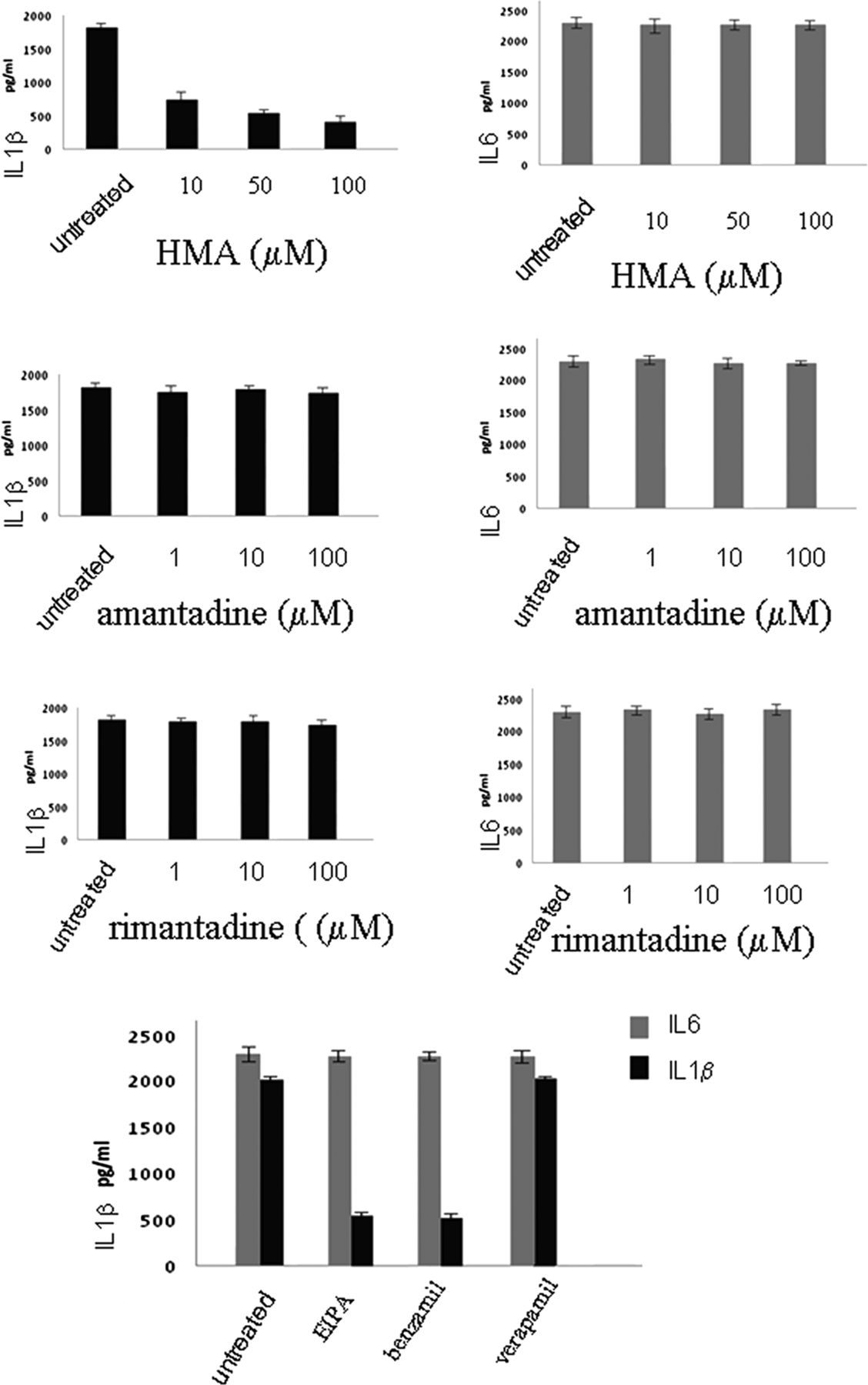
To investigate the mechanism of SH-induced inflammasome activation,10 we infected cells with RSV or with rgRSV-GF, and used drugs that block the ion channel of several other viroporins, such as amantadine and rimantadine which block the influenza M2 channel, and HMA which blocks the HIV Vpu channel as well as the p7 channel of Hepatitis C virus to obtain further understanding of the channel properties of SH protein (figure 4)
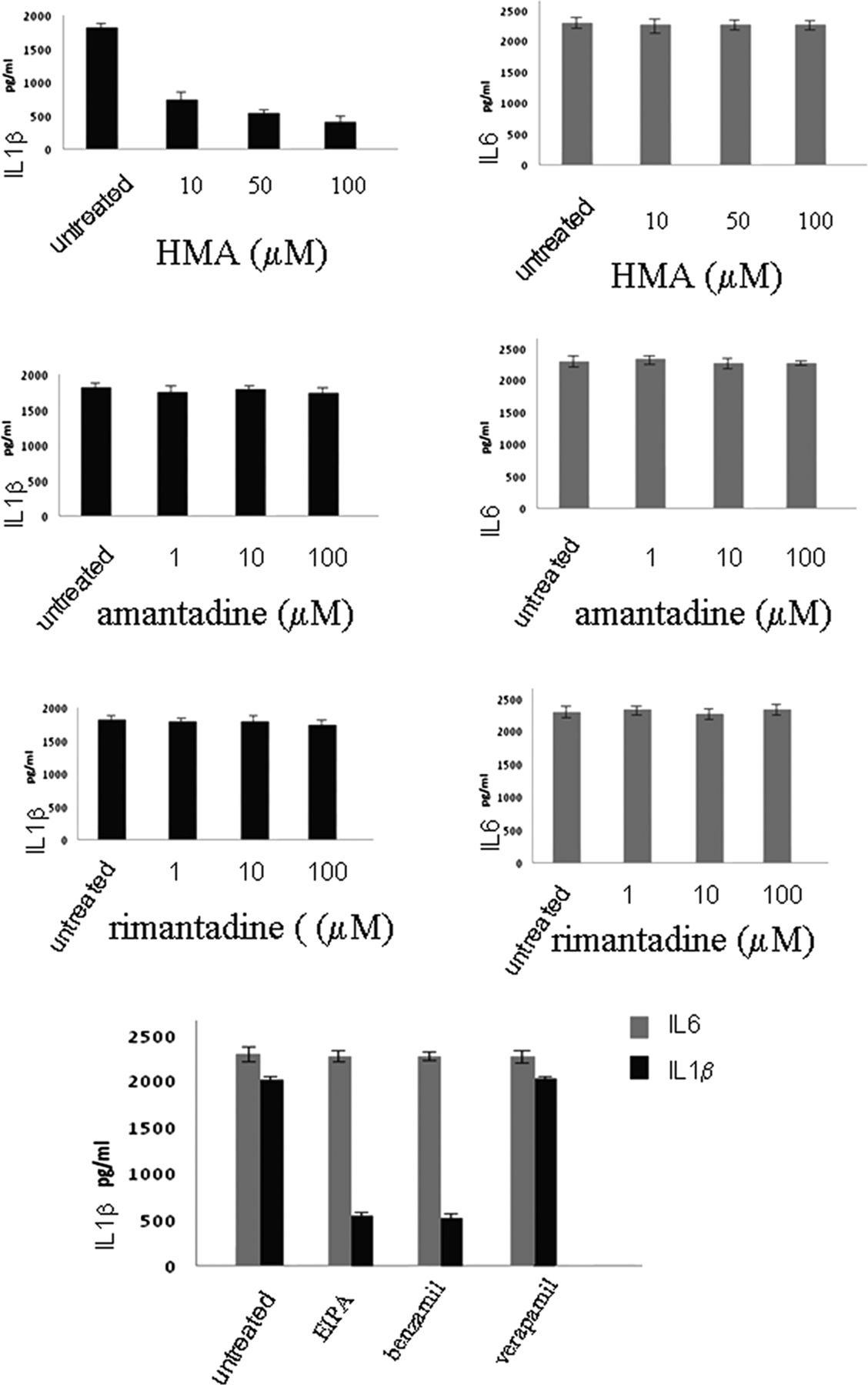
Effects of ion channel inhibitors on inflammasome activation. Lung epithelial cells were infected with respiratory syncytial virus and cultured in the presence or absence of hexamethylene amiloride or amantadine or rimantadine or EIPA, or benzamil or verapamil for 12 h. Supernatants were collected and tested for IL1β and IL6 secretion using the CBA bead array system (BectonDickinson). Fluorescence was detected using a FACSCalibur (BectonDickinson) The data presented are the mean of three independent experiments.
HMA was successful in inhibiting IL1β secretion upon RSV infection. However, there was no downregulation when amantadine or rimantadine were used. Having observed the effect of HMA, we then examined the effect of other ion channel inhibitors: EIPA and Benzamil, which also block Na+/H+ ion channels, and Verapamil, which blocks Ca+ channels. Verapamil had no effect on IL1β secretion, whereas HMA, EIPA and Benzamil reduced IL1β secretion in RSV infection. These results suggest that SH displays is selective for monovalent cations (Na+ and K+) similar to the Hepatitis C virus (HCV) p7 protein (figure 4).
SH interactions
To elucidate the intracellular mechanism by which SH activates NLRP3 inflammasome, we examined SH trafficking in the cell. Previous studies have shown that RSV assembly and SH accumulation occur within lipid raft structures on the cell surface and into lipid raft domains in the Golgi network.19
Lung cells were infected with RSV fixed and permebealised with 0.2% saponin. An anti-SH antibody previously described was used for SH labelling. The SH protein localised at the endoplasmic reticulum and Golgi complex membranes. Whereas NLRP3 was distributed in the cytoplasm and after RSV infection, much of the NLRP3 as well as ASC was found in the Golgi (figure 5).
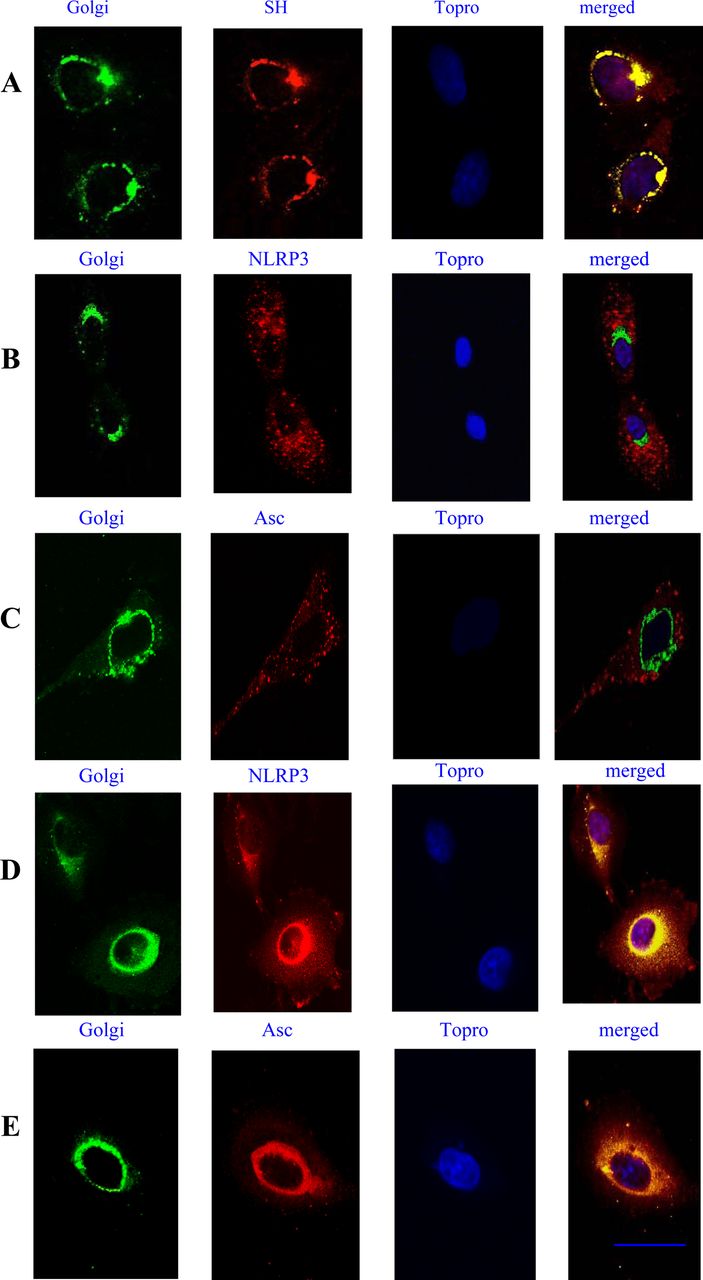
Localisation of small hydrophobic (SH), NLRP3 and ASC in the Golgi. Lung epithelial cells were stimulated with respiratory syncytial virus (RSV) for 6 h and then stained with GM130 mAb, followed by Alexa 488-Fab mouse-specific Ig to label the Golgi system. An anti-SH antibody was used to label the SH RSV protein followed by Alexa 546-Fab mouse-specific Ig. The nucleus was labelled using the nuclear stain Topro-3 (A). Ustimulated cells (B, C), or cells stimulated with RSV for 6 h (D, E), were stained for NLRP3 using a rabbit anti-NLRP3 Fab conjugated to Alexa 546, or a goat anti-ASC Fab conjugated to Alexa 546. The nucleus was labelled using the nulear stain Topro-3. Yellow regions indicate localisation of receptors. Cells were imaged using a Zeiss 510 confocal microscope. Bars 10 μm. The data presented are the mean ± SD of four independent experiments.
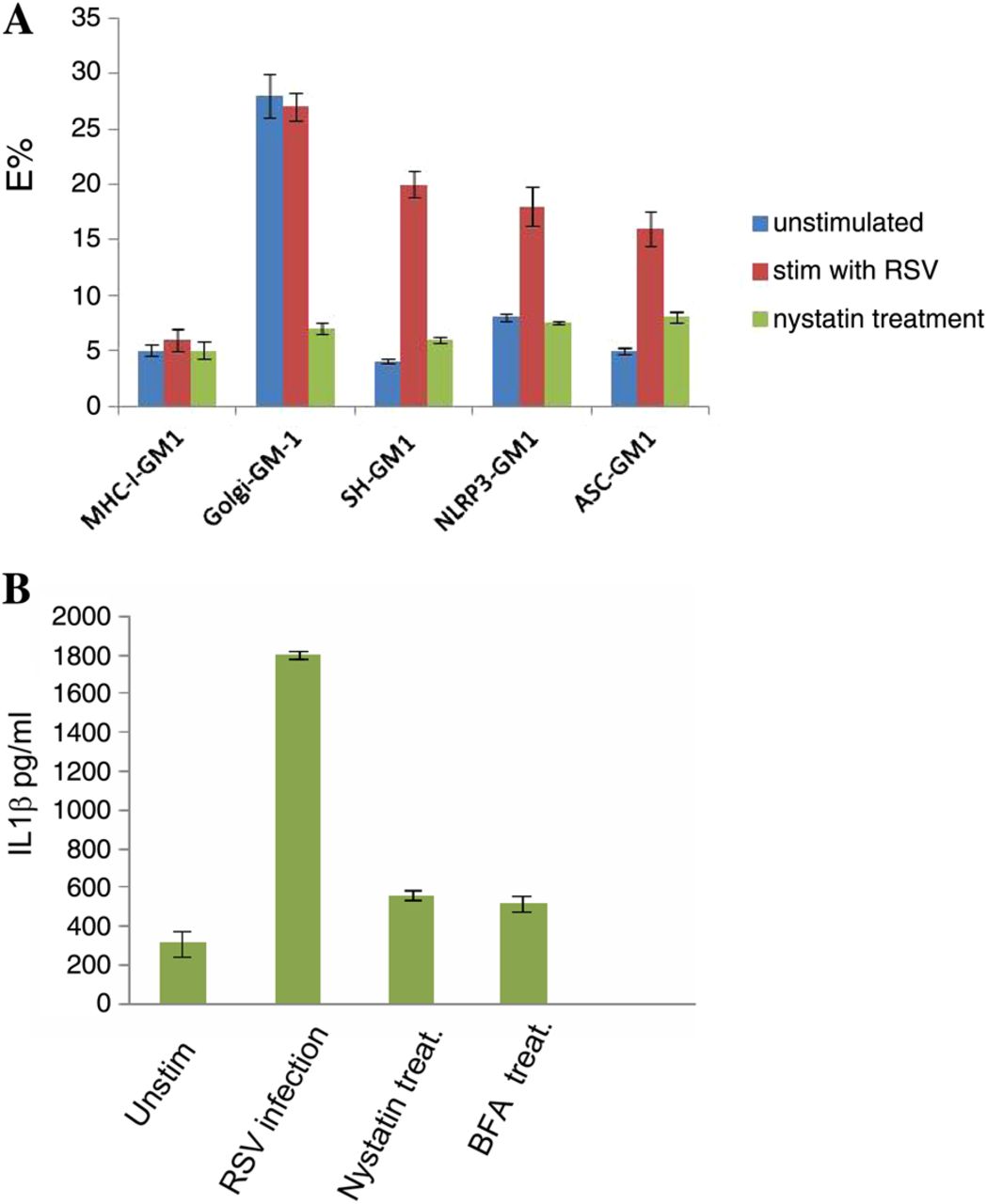
To analyse the role of lipid raft membranes within the Golgi complex in SH-induced inflammasome activation, cells were labelled with cholera toxin B subunit conjugated to TRITC (CTX-TRITC). CTX binds to the lipid raft marker GM1, and incubation at higher temperatures (between 25°C and 37°C) allows CTX to be internalised and transported to the Golgi.20 We used this method to label GM1-enriched lipid rafts within the Golgi compartment. SH was labelled with an SH antibody conjugated to Cy5, and used FRET to determine if the SH protein was in lipid rafts. We detected clear FRET signals in infected cells revealing associations between SH and lipid rafts, and between NLRP3 and lipid rafts (figure 6A). When cells were treated with nystatin, which disrupt caveolae and lipid rafts, the signal was lost. This result is consistent with NLRP3 and ASC redistribution that followed nystatin treatment of infected cells, showing that lipid raft disruption inhibited inflammasome activation since the redistribution of NLRP3 and ASC will inhibit their interactions and formation of inflammasome (figure 7). Furthemore, IL1β production in RSV-infected cells was dramatically reduced after nystatin treatment (figure 6B).

Lipid raft interactions with small hydrophobic (SH) and NLRP3. Fluorescence energy transfer fluorescence resonance energy transfer is a non-invasive imaging technique used in determining molecular associations. Lung epithelial cells were stimulated with respiratory syncytial virus (RSV) and cultured in the presence or absence of 25 μM nystatin. Lipid rafts were detected using Cholera toxin-TRITC to label GM1. The SH, NLRP3 or ASC were labelled with specific antibodies conjugated to Cy5. MHC-class-I interactions with GM1 were used as a negative control, whereas Golgi interactions with GM1 were used as a positive control. MHC-class-I was detected using W6/32 antibody conjugated to Cy5. Golgi was stained with GM130 mAb conjugated to Cy5 (A). Cells were imaged using a Zeiss 510 confocal microscope. Supernatants were also collected from lung epithelial cells stimulated with RSV and cultured in the presence or absence of 25 μM nystatin or 10 μM brefeldin A and tested for IL1β secretion using the CBA bead array system (BectonDickinson) (B). The data presented are the mean±SD of four independent experiments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NLRP3, ASC redistribution by nystatin treatment. Lung epithelial cells were stimulated with respiratory syncytial virus and then either left untreated (A) or cultured with 25 μM nystatin (B) or 10 μM brefeldin A (C). Golgi was stained with GM130 mAb followed by Alexa 488-Fab mouse-specific Ig. Cells were stained for NLRP3 using a rabbit anti-NLRP3 Fab conjugated to Alexa 546, and a goat anti-ASC Fab conjugated to Alexa 647. Cells were imaged using a Zeiss 510 confocal microscope. Bars 10 μm. The data presented are the mean±SD of four independent experiments.
In order to verify the importance of Golgi colocalisation in inflammasome activation, we utilised brefeldin A (BFA). The fungal metabolite BFA has been known to induce rapid and reversible disassembly of the Golgi stack into tubules and vesicles, resulting in the redistribution of Golgi-resident enzymes and accumulation of proteins in the endoplasmic reticulum (ER) in a reversible manner.21 ,22
Treatment with BFA disassembled the Golgi and resulted in NLRP3 and ASC redistribution (figure 7C) which blocked inflammasome activation by RSV since there was no IL1β production in the supernatant of cells treated with BFA (figure 6B).
Discussion
In this study, we have found that RSV is capable of triggering NLRP3 activation. This is in agreement with the recent study by Segovia et al. Our data clearly demonstrates that RSV is able to trigger NLRP3 inflammasome activation on its own, without the need for priming the cells with another PAMP as it has previously been proven necessary for other viruses, such as encephalomyocarditis virus and VSV.23 RSV appears to be able to provide both signal 1 and signal 2 for inflammasome activation on its own. The question that remains is: how are signals 1 and 2 triggered?
In order to answer this question, we initially investigated how signal 1 is triggered. There are many potential PRRs that might trigger signal 1 in the case of RSV, such as TLR4,12 TLR7,13 TLR3 and RIG-I.14 In order to investigate whether one or more of these PRRs trigger signal 1, we knocked down their expression without detecting reduction in IL1β secretion. Reduction in transcription of IL1β secretion was seen only when TLR4 was knocked down, suggesting that TLR4 is solely responsible for triggering signal 1 of RSV-induced inflammasome activation.
We have also knocked down TLR2; however, it had no effect in IL1β secretion. These findings are contrary to those by Segovia et al, who have reported that TLR2 triggers these responses. The differences observed in the two studies, might be due to the different cell types used. The study of Segovia et al was mainly performed on mouse knockout cells. Our study was performed in human lung epithelial cells. These cells are the main target of human RSV in vivo. In a mouse model, RSV might be detected by different PRRs.
Once we demonstrated that signal 1 is triggered by TLR4, we proceeded to investigate how signal 2 is triggered. Three models have been proposed. One model supports that extracellular ATP triggers K+ efflux24 and induces NLRP3 recruitment, another, the release of lysosomal contents caused by lysosome disruption25 ,26 and NLRP3 activation due to production of ROS.27
We examined RSV viral proteins that might enhance membrane permeability since it has been shown that the influenza M2 ion channel induces inflammasome activation by modulating the intracellular K+ concentration.15 The RSV SH protein has been classified as a viroporin. During the infection of cells by the virus, membrane permeability is modified by the use of viroporins which facilitate virus entry. The main activity of viroporins is to create pores at biological membranes to permit the passage of ions and small molecules; this is useful both in the entry as well as the exit mechanism of the virus.
In order to determine whether RSV SH protein contributes to NLRP3 activation, we used RSV mutants lacking the viroporin SH. These mutants have been shown in previous studies to grow and replicate efficiently in cell culture.18 Our results demonstrate that the RSV mutants lacking the viroporin SH are unable to trigger inflammasome activation, thus suggesting that signal 2 is triggered from the SH protein, possibly by the formation of a pore or channel on the plasma membrane. RSV SH protein joins an increasing list of viral proteins that have been shown to form ion channels in lipid bilayers, including picornavirus 2B,28 ,29 HIV-1 Vpu30 ,31 and HCV p7 which are permeable to both Na+ and K+ ions, and display relatively low ion selectivity,32 as well as M2 influenza viroporin which exhibits proton conductance and has been shown to activate the NLRP3 inflammasome.15
The use of drugs which block viral ion channels were shown to be able to inhibit IL1β secretion induced by RSV infection, suggesting that SH functions similar to HCV p7 which is selective for monovalent cations (Na+ and K+).
It has previously been shown that SH associates with lipid rafts on the cell surface, but after internalisation it accumulates in the Golgi complex within membrane structures that are enriched with the lipid raft marker, GM1.19 Our FRET studies confirmed that SH was indeed localised intracellularly in lipid rafts in the Golgi membranes. Interestingly, NALP3 was also shown to accumulate in the Golgi apparatus and to associate with the lipid raft marker GM1, upon RSV infection.
Lipid rafts are specialised membrane microdomains which provide a favourable environment for intramolecular cross-talk, but also aid in expediting signal transduction. Several new studies have found accumulation of ion channels and their modulators in lipid rafts,33 ,34 which is in agreement with our findings. Lipid rafts are not only on the plasma membrane, but have been shown to accumulate in the Golgi network as well. Their function there is believed to be the sorting of secretory cargo,35 and in the case of RSV, it might be taking advantage of that for its exit mechanism.
Our data show that upon infection with RSV, the SH protein is targeted to the Golgi where it must function as an ion channel. In addition, there is trafficking of NLRP3 from the cytoplasm to the Golgi where it localises within lipid rafts structures there. These intracellular raft structures seem to be crucial for the accumulation of ion channels and their modulators. Sensing of cellular stress imposed by imbalances in ionic concentrations in intracellular vesicles could be a general viral recognition pathway that can be utilised by the infected host cell to signal the activation of NLR inflammasomes.
Acknowledgments
This work was supported by a grant awarded from the Children and Young People's Research Network (NISCHR, Wales). The authors would like to thank Drs Mark Peeples and Peter Collins for the rgRSV and rgRSV-GF viruses, as well as critical reading of the manuscript, and Dr Biao He for the antiserum to the SH protein.
References
Footnotes
-
Contributors Conception and design (KT, MT, SK), acquisition of data, or analysis and interpretation of data (KT, MT, SK, EV), drafting the article or revising it critically for important intellectual content (KT, MT, SK, EV).
-
Funding Children & Young People's Research Network (NISCHR, Wales).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Respiratory infection
- Chest clinic
- Airwaves