Article Text

Abstract
We present a case of a 32-year-old woman who was diagnosed with lymphangioleiomyomatosis (LAM) after detecting a mass in the upper anterior mediastinum. Two years after presentation another metastatic localisation of LAM occurred in the cervical region. With this article we would like to highlight the fact that there are still a lot of unanswered questions, especially regarding the best management of extrapulmonary LAM.
- Extrapulmonary
- lymphangioleiomyomatosis
- mediastinum
- management
Statistics from Altmetric.com
Case report
Aurélie Derweduwen, MD (AD): We were asked to review a 32-year-old female suffering from intermittent haemoptysis combined with progressive dyspnoea. Apart from a history of pneumonia she had never been sick. She had a negligible smoking history. Bronchoscopy was normal and pulmonary embolism had been excluded. A chest CT scan was performed and a thymus mass was identified (figure 1). This mass was subsequently removed by thoracoscopic surgery, complicated by chylothorax and a fixed high position of the left diaphragm. On histopathology it was diagnosed as a smooth muscle tumour of uncertain malignant potential. As initial symptoms persisted patient was referred to guide further diagnosis and management. Patient suffered from exertional dyspnoea (NYHA II), thoracic cage pain, intermittent haemoptysis and low back pain. Detailed clinical examination revealed no abnormalities in the cardiorespiratory system, abdomen or pelvis. High Resolution CT revealed multiple bilateral small cysts with well-defined borders and uniform size, diffusely involving the lungs and not sparing the lung bases (figure 2). An abdominal CT showed solid and cystic retroperitoneal lymphadenopathies. Pulmonary function test showed a slight reduction in forced expiratory volume in one second (FEV1) (2.16 l or 70% predicted) and DLCO of 40% predicted. Patient was taking low dose paracetamol and oral progesterone (0.075 mg once daily).

Chest CT scan identified a thymus mass.
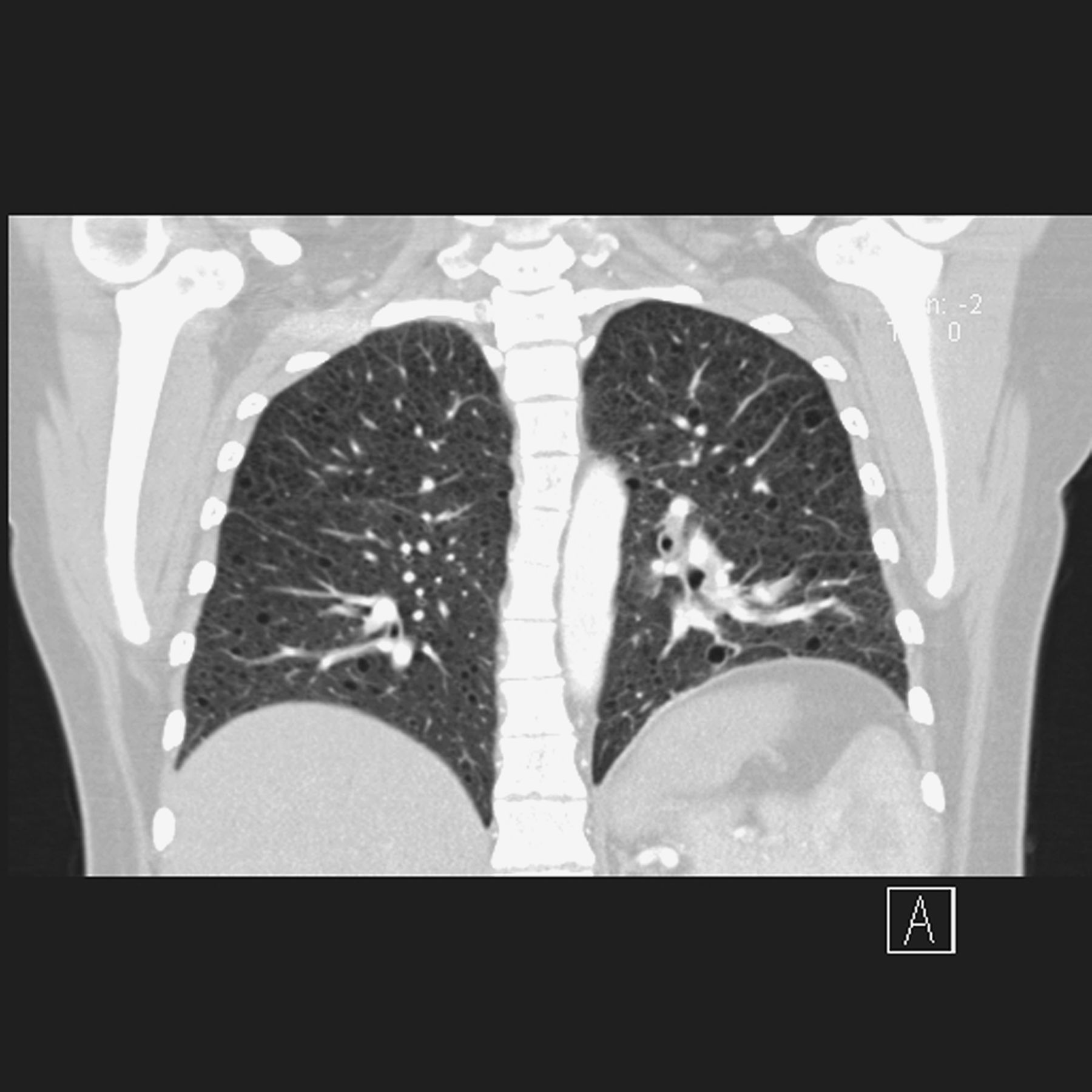
HRCT revealed multiple bilateral small cysts.
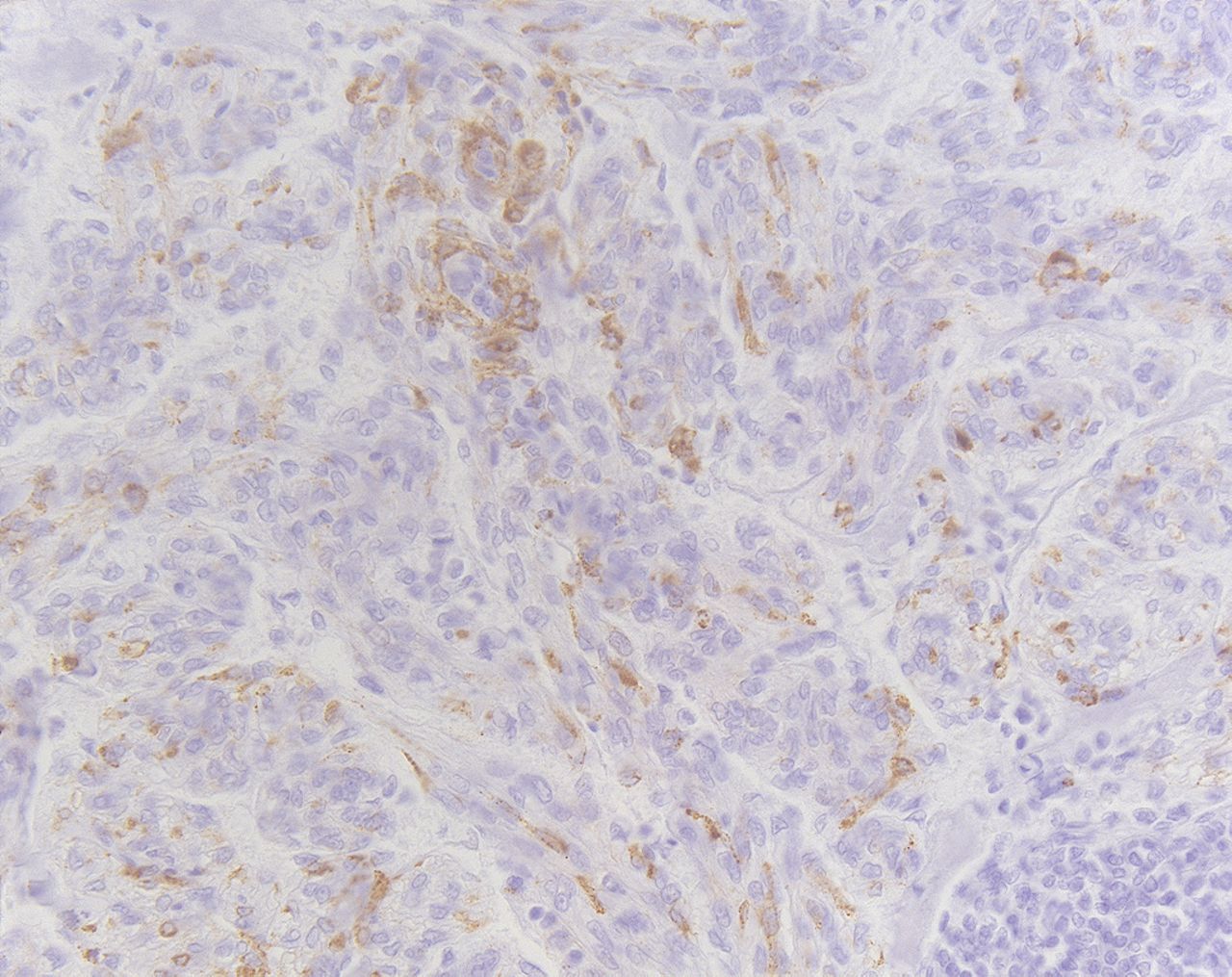
Wim Wuyts, MD, PhD (WW): The resection piece of the so-called smooth muscle tumour of uncertain malignant potential around the thymus was reviewed by our pathologist. Besides preserved thymic tissue a multilobular mass with focal haemorrhagic areas was observed, but no major cystic spaces were seen. There was no necrosis and the tumour was not encapsulated. Histologically the tumour consisted of proliferating immature smooth muscle cells invading the thymus as well as the surrounding mediastinal adipose tissue, up to four mitosis per 10 high power fields were noted. Focally haemorrhagic clefts were present but no large cystic spaces. Immunohistochemistry for smooth muscle actin, desmin and HMB45 were positive (figure 3). Diagnosis of lymphangioleiomyomatosis LAM with an extrapulmonary manifestation was based upon history of dyspnoea, haemoptysis, characteristic lung high resolution CT, retroperitoneal lymph node involvement, thoracic chylous effusion and histopathology. The clinical course of LAM is highly variable and it is difficult to predict the prognosis of individual patients. DLCO and FEV1 are likely to be the best current indicators of disease progression, response to treatment and survival. LAM patients generally develop progressive airflow obstruction, intermittent pneumothorax, chylous collections or other complications. Although most patients with renal angiomyolipomas do not have symptoms, these tumours may bleed which can be life-threatening. The recent European Respiratory Society Task Force guidelines offer a good view of the best management in these patients. As LAM is rare, there are no proven therapies for LAM, and therefore management must be based on clinical experience. One quarter of the patients with LAM may have some improvement with β2 agonist inhalation. Other measures including smoking cessation, pulmonary rehabilitation, oxygen and prophylactic vaccinations remain important. Because LAM occurs almost exclusively in young women and as exacerbations seem to be related to pregnancy, exogenous oestrogens and contraceptive pills, it has been suggested in the past that oestrogens may play a role in the development or progression of LAM and should be avoided. Despite this recommendation there is no evidence for anti-oestrogen interventions (oophorectomy, tamoxifen, gonadotrophin releasing hormone agonists). Despite its widespread use there are no randomised placebo controlled trials supporting a role for the use of progesterone. It is possible that progesterone therapy does slow the progression of the disease, only based on case reports, therefore it should not be used routinely. In patients with a rapid decline, intramuscular progesterone may be tried.1

Immunohistochemistry for HMB45 was positive.
AD: After discussing the possible therapies with the patient we only used progesterone in contraceptive dose and did not augment its daily dose. Overall there is an absence of adequate therapeutic options and patients should be considered for lung transplantation if severe decline in lung function impairment or exercise capacity is reached.
Two years after the initial presentation, she complained of a sore feeling with paraesthesia and reduced force in the left lower arm. Physical examination revealed a painful solid swelling of the neck and the supraclavicular region with a 5 cm diameter. Further careful clinical examination showed no other abnormalities. Neurological examination retained slight sensory failure of the left lower arm, compatible with minimal plexus compression. An MRI showed a T2 hyperintense conglomerate mass of cystic lesions in the left cervical region (figure 4). The lesion extended to the upper mediastinum, surrounded the jugular vein and the arteria carotis communis and lay in close encounter to the plexus brachialis. No lymphadenopathies and no compression of the great vessels were seen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
MRI showed a T2 hyperintense conglomerate mass of cystic lesions in the left cervical region.
WW: Our patient was admitted to perform a biopsy and removal of the mass to prevent further neurological damage. This tissue biopsy was compared with the previous resection piece and showed identical histopathological findings. In contrast with the mediastinal mass this biopsy was predominantly cystic. Cysts were filled with chylous fluid. Further an identical proliferation of immature smooth muscle cells was present. The latter had the same immunohistochemical profile as the mediastinal tumour, this confirming the diagnosis of LAM. The size of extrapulmonary LAM lesions is determined by the presence of cysts containing chylous fluid, rather than proliferation of LAM cells.2
AD: One month after resection she complained of intermittent swelling of the same region without overt neurological symptoms. There was, however, an increased need for pain relief and an increase of dyspnoea (NYHA III). A new MRI showed reduction of the total number of cysts but swelling of the residual cysts. Lung function demonstrated a mild obstructive function (FEV1 60% predicted) and a slight further reduction of DLCO (37% predicted). During the next 6 months there was intermittent swelling of the supraclavicular and cervical region with increased pain of left arm, neck and head. Thanks to analgetics (paracetamol 1 g four times a day, tramadol hydrochloride 150 mg three times a day, oxycodon hydrochloride 5 mg once daily) the pain was rather controlled. Exertional dyspnoea became more obvious and lung function evaluation showed further decline. Daily activities were becoming more difficult despite rehabilitation and haemoptysis was becoming more frequent. Patient was evaluated for lung transplantation and activated on the waiting list. Therapy remained unchanged.
WW: An improved understanding of the molecular pathogenesis of the disease has identified several promising molecular targets for clinical trials. LAM is associated with mutations in the TSC2 or TSC1 genes. Consequently the regulation of a key protein kinase, mammalian target of rapamycin (m TOR), is defect. Therefore an inappropriate activation of the m TOR signalling pathway leads to cellular growth and lymphangiogenesis. Sirolimus, also called rapamycin (an m TOR inhibitor) blocks this pathway and restores the homeostasis in the LAM cell. A recent study indicated that sirolimus may be useful in selected patients with moderately severe LAM and rapidly declining lung function. Given the usually slow progression rate of LAM and the risks of sirolimus therapy, decisions should be made on an individual basis. Another study showed sustained regression of asymptomatic renal angiomyolipomas by continuation sirolimus for 2 years.1 ,3–5 We chose not to start with sirolimus as the patient was activated on the waiting list for lung transplantation. Sirolimus is known to increase the risk of invasive infections and to decrease wound healing, leading to anastomotic complications following lung transplantation. Surgical resection does not seem useful for extrapulmonary lymphangiomyomas as cysts may return after time.
AD: LAM is an uncommon lung disease and literature is sparse. Subjects with extrapulmonary LAM have been described in detail only in a small number of individual case reports, but in the last decade several centres assembled their LAM cases and reported their experiences. Extrapulmonary manifestation as the primary presentation in LAM is unusual and only accounted for five of the 50 cases described in a recent British series. Two patients presented with a pelvic lymph node mass, three with a renal tumour. In five patients there was a subcutaneous swelling, two in the abdominal wall and one in the buttock, upper thigh and neck. The swelling occurred intermittently in four patients, two of them had a lymphangiomyoma. The other swellings were explained by chylous effusion, ascites or both.6 In 2000, Matsui et al reviewed 22 cases of extrapulmonary LAM out of a series of 188 LAM patients. Most of the lesions of extrapulmonary LAM occurred in lymph nodes along the lymphatic vessels of the posterior mediastinum, the upper retroperitoneal area and the pelvic cavity. None of these patients had supraclavicular or cervical localisation of LAM, but two patients had extrapulmonary LAM in the upper mediastinum. Overall these masses were well defined, encapsulated and ranged from 1 to 20 cm in greatest dimension. The masses greater than 3 cm had prominent cysts and were filled with chylous fluid. It was surprising that in 13 of the 17 patients with available follow-up data the diagnosis of extrapulmonary LAM preceded that of pulmonary LAM.2 In a French review of 69 LAM patients the initial symptoms were extrathoracic in 10 patients. Renal angiomyolipoma was diagnosed in seven patients before LAM diagnosis. Mediastinal lymphadenopathy was rare (9%) or absent.7
WW: Except for angiomyolipoma the European Respiratory Society Task force did not describe management of extrapulmonary LAM, which remains a subject of discussion. It is time to initiate new initiatives to pool the rare cases of extrapulmonary LAM and to initiate new clinical trials. Meanwhile, best supportive care and finally lung transplantation seem the only valid approaches.
Footnotes
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.