Article Text

Abstract
Background 20–30% of patients with asthma have neutrophilic airway inflammation and reduced responsiveness to steroid therapy. They often have chronic airway bacterial colonisation and Haemophilus influenzae is one of the most commonly isolated bacteria. The relationship between chronic airway colonisation and the development of steroid-resistant neutrophilic asthma is unclear.
Objectives To investigate the relationship between H influenzae respiratory infection and neutrophilic asthma using mouse models of infection and ovalbumin (OVA)-induced allergic airways disease.
Methods BALB/c mice were intratracheally infected with H influenzae (day 10), intraperitoneally sensitised (day 0) and intranasally challenged (day 12–15) with OVA. Treatment groups were administered dexamethasone intranasally during OVA challenge. Infection, allergic airways disease, steroid sensitivity and immune responses were assessed (days 11, 16 and 21).
Results The combination of H influenzae infection and allergic airways disease resulted in chronic lung infection that was detected on days 11, 16 and 21 (21, 26 and 31 days after infection). Neutrophilic allergic airways disease and T helper 17 cell development were induced, which did not require active infection. Importantly, all features of neutrophilic allergic airways disease were steroid resistant. Toll-like receptor 4 expression and activation of phagocytes was reduced, but most significantly the influx and/or development of phagocytosing neutrophils and macrophages into the airways was inhibited.
Conclusions The combination of infection and allergic airways disease promotes bacterial persistence, leading to the development of a phenotype similar to steroid-resistant neutrophilic asthma and which may result from dysfunction in innate immune cells. This indicates that targeting bacterial infection in steroid-resistant asthma may have therapeutic benefit.
- Chronic infection
- Haemophilus influenzae
- neutrophilic asthma
- steroid resistance
- innate immune dysfunction
- Th17
- asthma
- bacterial infection
- innate immunity
- cytokine biology
- eosinophil biology
- exhaled airway markers
- neutrophil biology
- airway epithelium
- bronchiectasis
- complementary medicine
- cystic fibrosis
- lung cancer
- non-small cell lung cancer
- paediatric lung disaese
- rare lung diseases
- tobacco and the lung
- asthma pharmacology
- COPD exacerbations
- COPD pathology
- COPD pharmacology
- macrophage biology
- respiratory infection
- asthma guidelines
- asthma mechanisms
- cough/mechanisms/pharmacology
- COPD mechanisms
- allergic lung disease
- paediatric asthma
- pneumonia
Statistics from Altmetric.com
- Chronic infection
- Haemophilus influenzae
- neutrophilic asthma
- steroid resistance
- innate immune dysfunction
- Th17
- asthma
- bacterial infection
- innate immunity
- cytokine biology
- eosinophil biology
- exhaled airway markers
- neutrophil biology
- airway epithelium
- bronchiectasis
- complementary medicine
- cystic fibrosis
- lung cancer
- non-small cell lung cancer
- paediatric lung disaese
- rare lung diseases
- tobacco and the lung
- asthma pharmacology
- COPD exacerbations
- COPD pathology
- COPD pharmacology
- macrophage biology
- respiratory infection
- asthma guidelines
- asthma mechanisms
- cough/mechanisms/pharmacology
- COPD mechanisms
- allergic lung disease
- paediatric asthma
- pneumonia
Key messages
What is the key question?
How is chronic bacterial infection associated with steroid-resistant neutrophilic asthma.
What is the bottom line?
The combination of infection and allergic airways disease induces the complete suppression of phagocytic cells in the lung that leads to chronic infection, neutrophilic inflammation and steroid resistance.
Why read on?
This indicates how bacterial infections may be associated with steroid-resistant neutrophilic asthma.
Introduction
Asthma is a chronic inflammatory condition of the airways, frequently characterised by abnormal immune responses to environmental antigens, leading to recurrent episodes of cough, wheezing and breathlessness.1 Patients with neutrophilic asthma make up between 20% and 30% of patients with asthma, and are characterised by substantial increases in airway neutrophils. Their disease symptoms and neutrophilic inflammation are poorly responsive to corticosteroids, the mainstay of asthma therapy, and it has been suggested that airway neutrophilia may be a consequence of steroid treatment.2 ,3 Neutrophilic airway inflammation is also common to many obstructive airway diseases, including neutrophilic asthma, chronic obstructive pulmonary disease (COPD) and bronchiectasis.2 ,4 ,5
The cellular innate immune system of the respiratory tract includes epithelial cells, neutrophils, macrophages and dendritic cells. It is considered the first line of defence against invading pathogens, mounts an immediate non-specific response, and promotes antigen-specific responses by the adaptive component.6 Defects in innate immunity may be important in the development of steroid-resistant neutrophilic asthma, and increased sputum neutrophils, interleukin (IL)-8 levels and expression of toll-like receptors (TLRs) have been demonstrated.4 ,7
Neutrophils and macrophages have critical roles in innate immunity and are important contributors to airway inflammation. Therefore, dysfunction in these cells may lead to the development or worsening of steroid-resistant neutrophilic asthma. Airway neutrophils from patients with asthma are less activated compared with healthy controls, which may lead to impaired local defence and increased susceptibility to infections.8 Indeed, in COPD, which is also underpinned by neutrophilic bronchitis, airway neutrophils have impaired chemotaxis, and alveolar macrophages have reduced TLR2 expression.9 ,10
Infections by viruses and bacteria have pathogenic roles in asthma and induce asthma exacerbations.11–14 Chronic bacterial infection occurs frequently in airway diseases such as COPD, and bronchiectasis, and may be important in steroid-resistant neutrophilic asthma.7 ,15 ,16 In neutrophilic asthma H influenzae is one of the most commonly isolated bacteria and we have recently shown that infection in allergic airways disease drives a neutrophilic phenotype that is dependent on IL-17.7 ,15 ,17 However, the relationship between chronic H influenzae infection and the development of steroid-resistant neutrophilic asthma is not understood.
In this study we employed murine models of H influenzae infection and OVA-induced allergic airways disease to investigate if and how the combination of infection and allergic airways disease may interact to promote chronic infection and the development of steroid-resistant neutrophilic asthma.
Methods
See online supplement for additional details.
Experimental protocols
Female BALB/c mice, 6–8 weeks old, were inoculated intratracheally with 5×105 colony-forming units (CFU) non-typeable H influenzae (NTHi-289, in 30 μl phosphate-buffered saline) or 5×105 CFU ethanol-killed NTHi-289 and allergic airways disease was induced 10 days later (figures 1A and 2A).18 Mice were sensitised to OVA (50 μg, intraperitoneal injection, Sigma-Aldrich, Sydney, Australia), with Rehydrogel (1 mg, Reheis, Berkeley Heights, New Jersey, USA) in saline (200 μl), and subsequently challenged intranasally with OVA (10 μg/50 μl saline) on days 12–15 to induce allergic airways disease. Saline control groups received saline sensitisation with Rehydrogel by intraperitoneal injection and subsequent OVA challenges on days 12–15.19 Infection, immune responses and allergic airways disease were assessed on days 11, 16 and 21.

Allergic airways disease during Haemophilus influenzae infection promotes chronicity of infection. (A) Mice were inoculated intratracheally with 5×105 colony-forming units (CFU) of H influenzae, then 10 days later sensitised intraperitoneally (day 0) and challenged intranasally (days 12–15) with ovalbumin (OVA). Controls received saline sensitisation and OVA challenge. Allergic airways disease, immune responses and infection were assessed (days 11, 16 and 21) and compared with controls. (B) The effects of allergic airways disease on H influenzae recovery from lungs and bronchoalveolar lavage fluid combined were characterised. Data represent the mean ± SEM from six to eight mice. Significant differences between infected non-allergic (H influenza (Hi)) and infected allergic (Hi/OVA) groups are shown as ***p<0.001.

Killed Haemophilus influenzae (KHi) suppresses hallmark features of eosinophilic allergic airways disease. (A) Mice were administered with ethanol-killed H influenzae then sensitised (day 0) and challenged (days 12–15) with ovalbumin (OVA). The effects of KHi in allergic airways disease on the numbers of (B) total cells, (C) eosinophils and (D) neutrophils in bronchoalveolar lavage fluid (BALF) and OVA-induced (E) interleukin (IL)-5, (F) IL-13 and (G) interferon γ (IFNγ) release from MLN T-cell were assessed. Airway hyperresponsiveness measured as (H) dynamic compliance and (I) transpulmonary resistance in response to increasing doses of methacholine was determined. Significant differences between allergic (OVA) and non-allergic (saline) groups are shown as ###p<0.001, ##p<0.01, #p<0.05. Significant differences between infected allergic (H influenzae/OVA) and uninfected allergic (OVA) groups are shown as ***p<0.001, **p<0.01. Significant differences for resistance and compliance are for the entire dose–response curve.
Bacterial recovery
Serial dilutions of lung homogenates and bronchoalveolar lavage fluid (BALF) were plated onto chocolate agar plates (Oxoid, Thebarton, Australia) and incubated overnight (37°C, 5% CO2). Colonies were enumerated and bacterial numbers in lungs and BALF were determined and combined.17
Cellular inflammation
BALF cytospins and blood slides were stained with May-Grunwald Giemsa and differential leucocyte counts obtained using light microscopy.19
T-cell cytokines
Mediastinal lymph node (MLN) T-cell cultures were re-stimulated with OVA or ethanol-killed H influenzae and cytokine concentrations in supernatants assessed by ELISA.20
Lung cytokine expression
IL-13 and interferon γ (IFNγ) expression was determined relative to the reference gene hypoxanthine-guanine phosphoribosyltransferase in TRIZOL-extracted RNA from whole lung tissue according to manufacturer's instructions (Invitrogen, Mount Waverly, Australia).19 Primers used were hypoxanthine-guanine phosphoribosyltransferase Fwd 5′-aggccagactttgttggatttgaa-3′, Rev 5′-caacttgcgctcatcttaggcttt-3′; IL-13, Fwd 5′-aaacatgagtccagggagagcttt-3′, Rev 5′-actgagcttcccagatcacagagg-3′; IFNγ, Fwd 5′-gctcatcttaaggccagact-3′, Rev 5′-atttgaatccagggatccgaatt-3′.
Lung function
Airway hyperresponsiveness (AHR) was measured in response to increasing doses of methacholine by whole body invasive plethysmography as previously described.18
Th2 and Th17 cells
Single-cell lung and MLN preparations were stimulated with phorbol 12-myristate 13-acetate (PMA, 0.1 μg/ml) and ionomycin (1 μg/ml) in the presence of Brefeldin A (8 μg/ml, Sigma-Aldrich). Cells were stained for surface markers CD3, CD4 (BD Bioscience, San Diego, California, USA), fixed, permeabilised and stained for intracellular IL-13 and IL-17A (eBioscience, San Diego, California, USA). Cells were analysed by flow cytometry (FACS Canto, BD Bioscience).19–21
Dexamethasone treatment
Dexamethasone (Sigma-Aldrich) was prepared in sterile saline (1 mg/kg) and administered intranasally (50 μl, days 13–15, figure 3A).

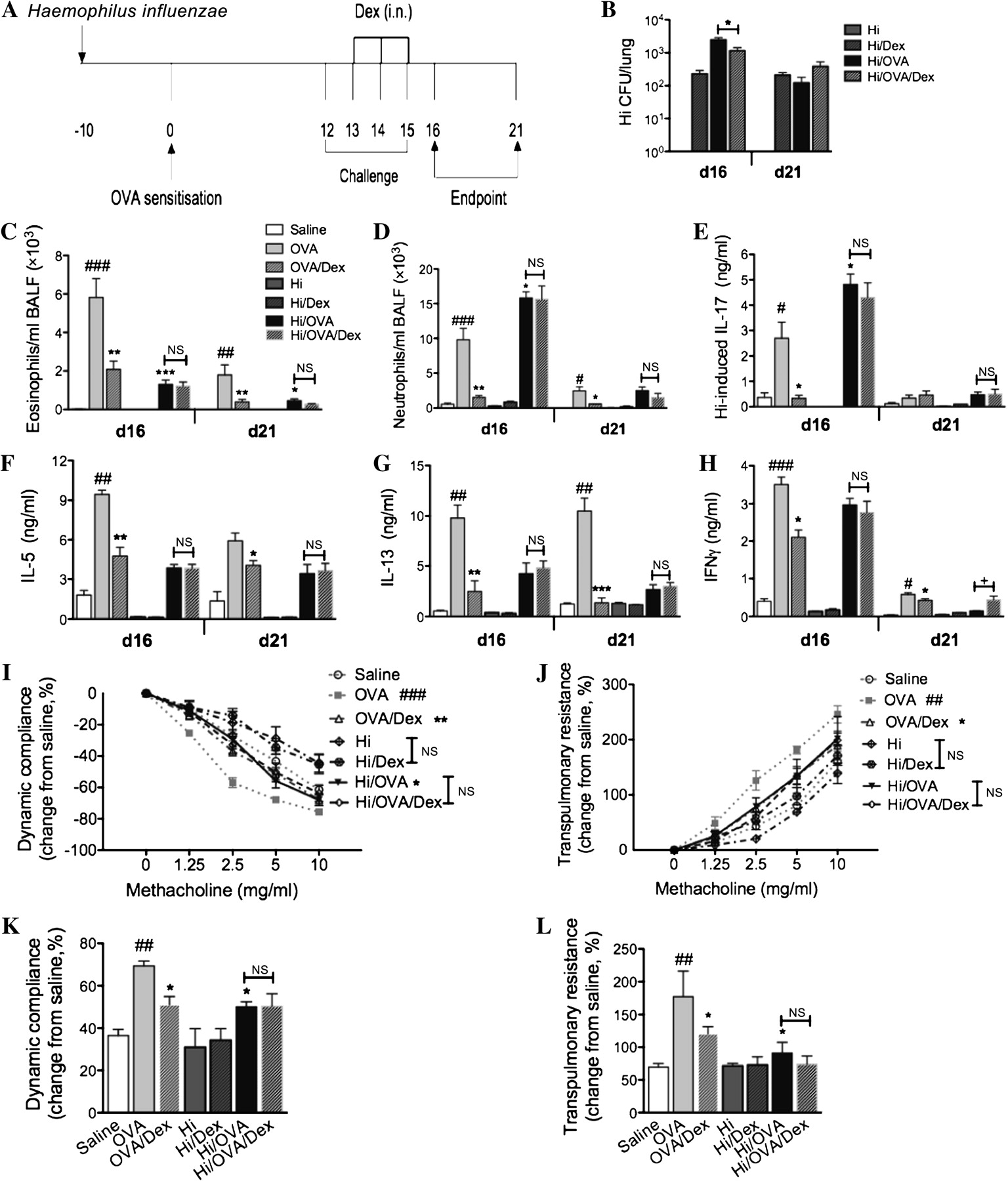
Chronic Haemophilus influenzae (Hi) infection in allergic airways disease induces steroid resistance. The effects of chronic Hi infection in allergic airways disease on responsiveness to steroid treatment were investigated. (A) Infected allergic groups were treated intranasally (i.n.) with dexamethasone (Dex) during ovalbumin (OVA) challenge (days 13–15) and allergic airways disease was assessed (days 16 and 21) and compared with controls. (B) To determine the effect of treatment on infection, bacterial recovery was assessed. To determine the effect of treatment on neutrophilic allergic airways disease, numbers of (C) eosinophils and (D) neutrophils in bronchoalveolar lavage fluid (BALF), (E) Hi-induced interleukin (IL)-17, OVA-induced (F) IL-5, (G) IL-13 and (H) interferon γ (IFNγ) were assessed. Airway hyperresponsiveness, measured as (I) dynamic compliance and (J) transpulmonary resistance in response to increasing doses of methacholine, was determined. Representative (K) compliance and (L) resistance data at 5 mg/ml are also shown. Data represent the mean ± SEM from six to eight mice. Significant differences between allergic (OVA) and non-allergic (saline) groups are shown as ###p<0.001, ##p<0.01, #p<0.05. Significant differences compared with uninfected allergic (OVA) groups are shown as ***p<0.001, **p<0.01, *p<0.05. Significant differences between untreated and treated groups are shown as +p<0.05. Significant differences for resistance and compliance are for the entire dose–response curve. NS, non-significant.
Mucus secreting cells
Lungs were imbedded in paraffin, sectioned (4–6 μm) and stained with periodic acid Schiff. Mucus secreting cell (MSC) numbers within the airway lumen (10×100 μm fields) were enumerated.20
Ciliary beat frequency
Ciliary beat frequency was measured for 1 min per section in tracheal transverse slices using transmitted light photometry and mean frequency (Hz) was calculated.22
Phenotyping of immune cells
Macrophages (CD11b+F4/80+, eBioscience) and neutrophils (CD11bhiGr-1hi, eBioscience) were stained for the activation marker CD62L, and TLR2 and TLR4 (eBioscience) and were analysed using flow cytometry.
Phagocytosis
Carboxyfluorescein succinimidyl ester (CFSE, 5 μM) labelled NTHi were incubated with BALF cells as previously described.23 BALF cells were stained for CD11b, F4/80 and Gr-1 and phagocytosis of CFSE-labelled NTHi was assessed using flow cytometry.
Oxidative burst
Lung cells were incubated with dihydroethidium (DHE, Sigma-Aldrich) for 15 min.23 Reactions were terminated by placing on ice, and cells were washed and fixed. Oxidative burst was assessed in fixed cells using flow cytometry.
Serum antibodies
Hi-specific immunoglobulin G1 (IgG1, using H influenzae as the capture antigen) was measured in serum by ELISA.21
Statistical analyses
Data are presented as mean ± SEM. Statistical significance for multiple comparisons was determined by one-way analysis of variance (ANOVA) with the Bonferroni post-test. One-way repeated measures ANOVA with Bonferroni post-test was used to analyse AHR data (GraphPad Prism Software, La Jolla, California, USA).
Results
Allergic airways disease during infection leads to chronic H influenzae infection
To investigate the relationship between infection and allergic airways disease, we first assessed the effect of allergic airways disease on H influenzae infection. In a recent study we showed that after inoculation with 5×105 CFU, H influenzae infection in the absence of allergic airways disease peaks after 5 days (1.56×106 ± 9×103 CFU), declines after 10 days (4×102 ± 1×102 CFU) and is undetectable by 16 days.17
Next, mice were inoculated with H influenzae and 10 days later, after the majority of H influenzae had been cleared, allergic airways disease was induced (figure 1A). The induction of allergic airways disease promoted the development of chronic infection with substantial levels of H influenzae recovered 21 (day 11 of the model), 26 (day 16 of the model) and 31 (day 21 of the model) days after inoculation of infected allergic (H influenzae/OVA) groups compared with infected non-allergic (H influenzae) controls (figure 1B). Uninfected allergic controls were also assessed and neither H influenzae nor commensal bacteria were detected (data not shown), ruling out the possibility that allergic airways disease induces substantial alterations in commensal species.
Chronic H influenzae infection in allergic airways disease suppresses eosinophilic inflammation
We have recently shown that infection during or after sensitisation to OVA suppresses eosinophilic allergic airways disease.17 To investigate if similar effects were induced by chronic H influenzae infection, mice were infected, allergic airways disease induced (figure 1A) and key features of allergic airways disease were assessed.
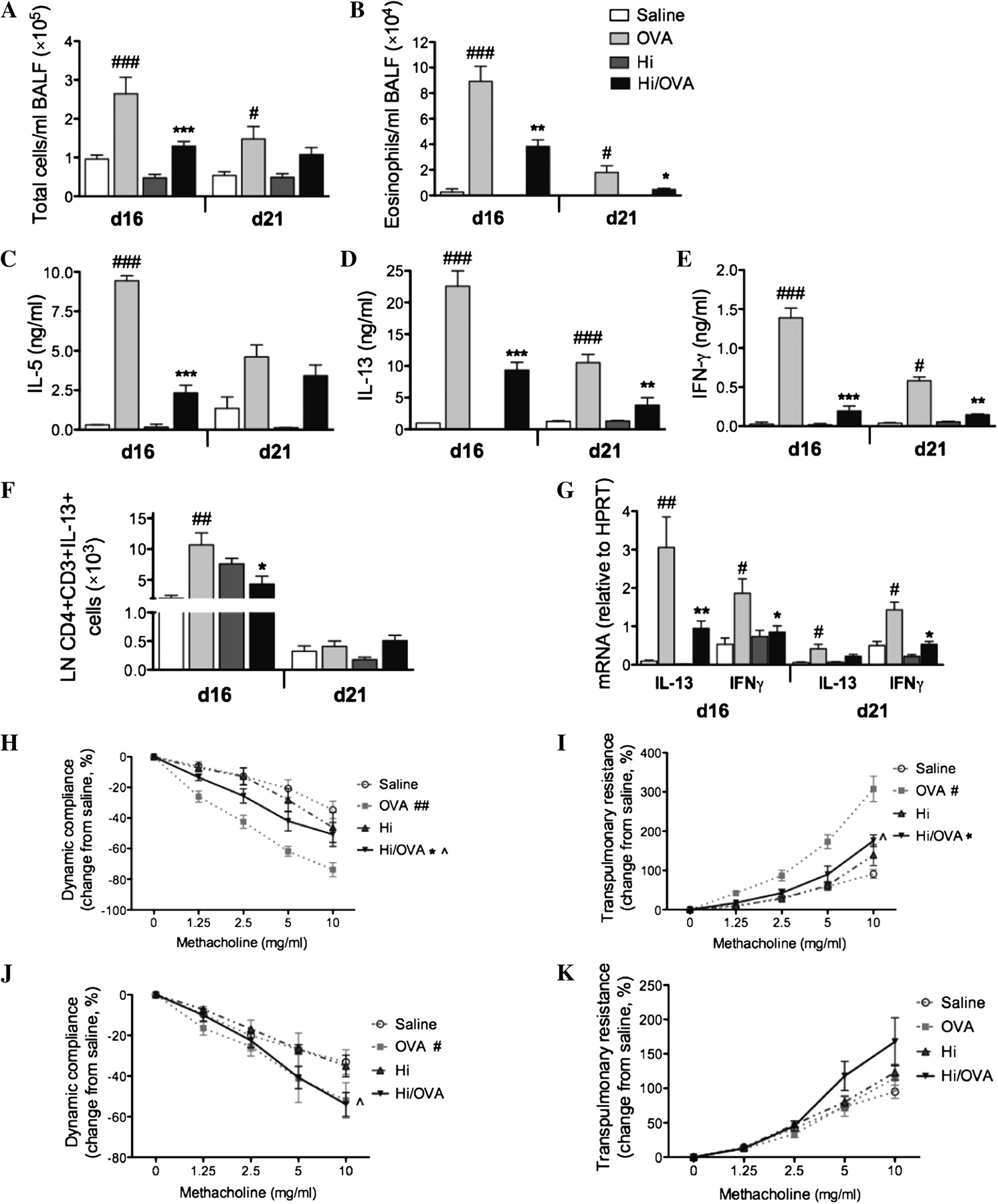
The induction of allergic airways disease (OVA groups) resulted in eosinophilic inflammation, OVA-induced IL-5, IL-13 and IFN-γ release from MLN T-cell and AHR (decreased dynamic compliance and increased transpulmonary resistance) compared with non-allergic (saline) controls (figure 4A–K). Infection alone had no effects on these features. However, infection during allergic airways disease significantly reduced the numbers of total cells and eosinophils in BALF compared with uninfected allergic (OVA) controls (figure 4A,B). The suppression of airway responses was accompanied by reductions in OVA-induced IL-5, IL-13 and IFN-γ release from T-cell (days 16 and 21), Th2 cells in the MLN (figure 4C–F) and expression of IL-13 and IFN-γ mRNA in the lungs at days 16 and 21 (figure 4G). Infection during allergic airways disease also significantly reduced AHR (increased airway dynamic compliance and reduced transpulmonary resistance), which however remained elevated compared with non-allergic controls. Specifically, in infected allergic (H influenzae/OVA) groups compared with uninfected non-allergic (saline) controls, dynamic compliance was reduced across the whole methacholine dose response curve on day 16 of the model (figure 4H) and at the highest methacholine dose (10 mg/ml) on day 21 (figure 4J). Transpulmonary resistance was also increased but was only statistically significantly so at the highest methacholine dose (10 mg/ml) on day 16 (figure 4I).

Chronic Haemophilus influenzae (Hi) infection suppresses hallmark features of eosinophilic allergic airways disease. The effects of chronic Hi infection in allergic airways disease on the numbers of (A) total cells and (B) eosinophils in bronchoalveolar lavage fluid (BALF) were assessed. The effects of infection on ovalbumin (OVA)-induced (C) interleukin (IL)-5, (D) IL-13 and (E) interferon γ (IFNγ) release from mediastinal lymph node T-cell, (F) mediastinal lymph node (LN) T helper 2 cells, and (G) IL-13 and IFNγ mRNA expression relative to hypoxanthine-guanine phosphoribosyl-transferase (HPRT) in the lungs were also assessed. (H, I) Airway hyperresponsiveness, measured as dynamic compliance and transpulmonary resistance in response to increased doses of methacholine, was determined on day 16 and (J, K) day 21, respectively. Significant differences between allergic (OVA) and non-allergic (saline) groups are shown as ###p<0.001, ##p<0.01, #p<0.05. Significant differences between infected allergic (Hi /OVA) and uninfected allergic (OVA) groups are shown as ***p<0.001, **p<0.01, *p<0.05. Significant differences in resistance and compliance are for the entire dose–response curve (H–K), although significant differences between infected allergic (Hi/OVA) and uninfected non-allergic (saline) groups for the whole curve or at 10 mg/ml methacholine (I, J) are shown as ∧p<0.05.
Chronic H influenzae infection in allergic airways disease induces neutrophilic inflammation and Th17 cell and IL-17 responses
H influenzae has frequently been isolated from patients with neutrophilic asthma who also have increased sputum neutrophilic inflammation. We have also recently shown that infection during or after sensitisation to OVA induces neutrophilic allergic airways disease that is dependent on IL-17.17 Therefore, we investigated whether chronic H influenzae infection in allergic airways disease also induced enhanced neutrophilic inflammation.
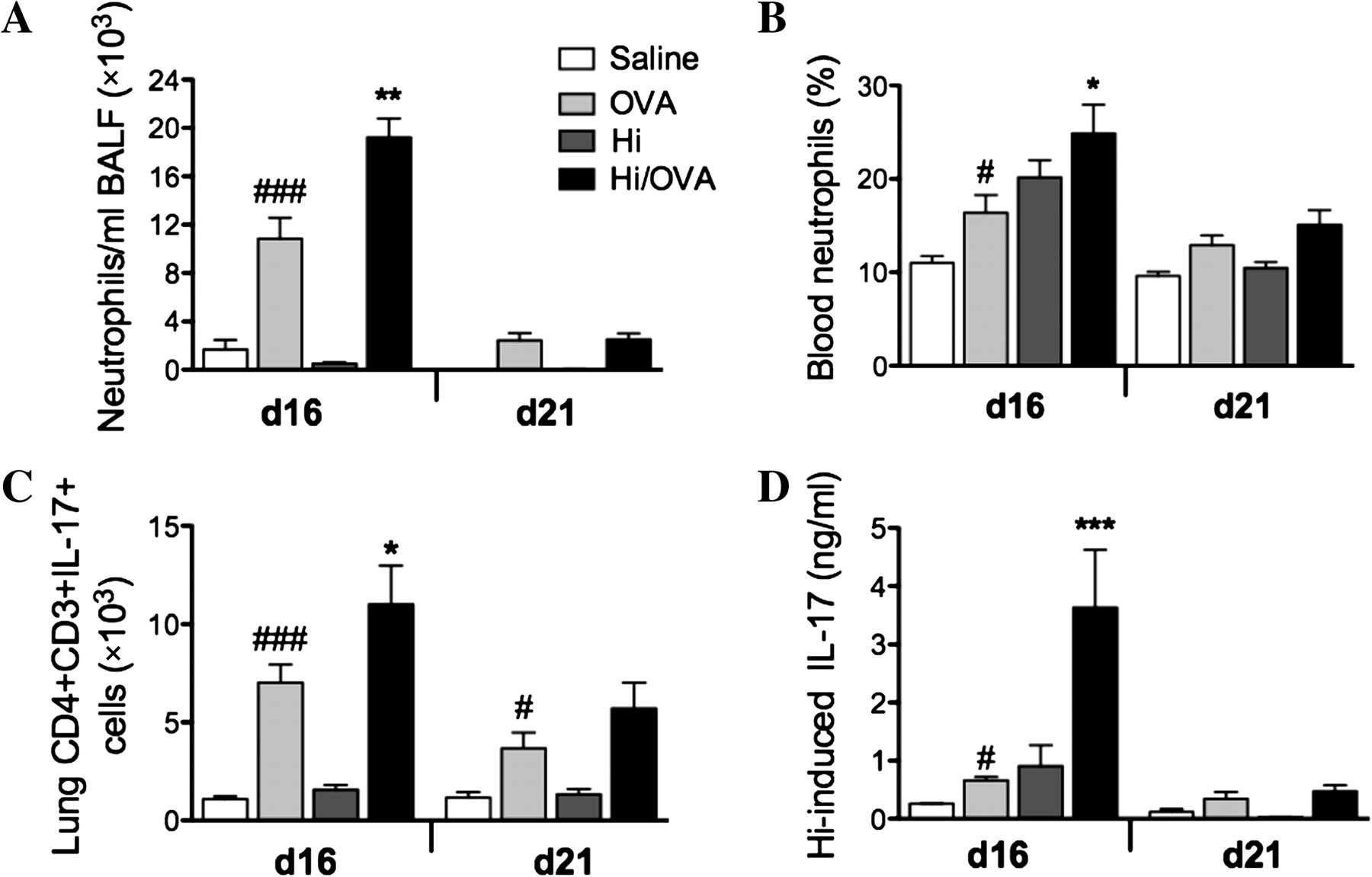
The induction of allergic airways disease was associated with mild neutrophilic inflammation (figure 5A,B). The combination of chronic infection and allergic airways disease significantly increased the influx of neutrophils into the airway and blood compared with uninfected allergic groups on day 16 (figure 5A,B).

Chronic Haemophilus influenzae (Hi) infection in allergic airways disease induces neutrophilic inflammation and T helper 17 responses. The effects of chronic Hi infection in allergic airways disease on the levels of neutrophils in (A) the airways and (B) blood, (C) Th17 cell numbers in the lung and (D) Hi-induced interleukin (IL)-17 release from mediastinal lymph node T-cell were assessed. Data represent the mean ± SEM from six to eight mice. Significant differences between allergic ovalbumin (OVA) and non-allergic (saline) groups are shown as ###p<0.001, #p<0.05. Significant differences between infected allergic (Hi/OVA) and uninfected allergic (OVA) groups are shown as ***p<0.001, **p<0.01, *p<0.05. BALF, bronchoalveolar lavage fluid.
Patients with asthma have increased sputum levels of IL-17 mRNA, and IL-17 stimulates the recruitment of neutrophils. Therefore, we assessed the numbers and levels of Th17 cells and IL-17 in infection and allergic airways disease. The induction of allergic airways disease increased the numbers of Th17 cells in lung tissue and IL-17 release from MLN T-cell stimulated with H influenzae on day 16 (figure 5C,D). Infection in allergic airways disease significantly increased the numbers of Th17 cells in the lung and IL-17 release from MLN T-cell (figure 5C,D). There were no differences in OVA-induced IL-17 production (data not shown). In other studies we used monoclonal antibody blocking studies to show that IL-17 is required for the development of the infection-induced neutrophilic inflammatory phenotype.18
Killed H influenzae suppresses hallmark features of eosinophilic allergic airways disease
To investigate whether an active infection was required for the induction of a neutrophilic phenotype, the effect of killed H influenzae on allergic airways disease was assessed. Mice were administered with 5×105 CFU of ethanol-killed bacteria and the effect of allergic airways disease was assessed on day 16 (figure 2A).
The combination of killed H influenzae and allergic airways disease (killed H influenzae/OVA) significantly decreased total cells, eosinophils and neutrophils in the BALF (figure 2B–D) and OVA-induced IL-13 and IFNγ release from T-cell compared with allergic controls (figure 2E–G). Killed H influenzae did not have any effect on AHR (figure 2H–I).
Chronic H influenzae infection in allergic airways disease induces steroid resistance
Many patients with neutrophilic asthma do not respond well to steroid therapy. Therefore, we investigated the sensitivity of infection and neutrophilic allergic airways disease to dexamethasone treatment.
Allergic and infected allergic groups were administered dexamethasone (1 mg/kg) intranasally during OVA challenge (days 13–15, figure 3A), and infection and allergic airways disease were assessed. Steroid treatment caused a small reduction in bacterial numbers in infected allergic (H influenzae/OVA/dexamethasone) groups at day 16 but not at day 21. Treatment significantly increased bacterial numbers in treated infected (H influenzae/dexamethasone) groups compared with untreated infected (H influenzae) controls at days 16 and 21 (figure 3B).
Dexamethasone treatment of uninfected allergic (OVA/dexamethasone) groups resulted in a significant reduction in all the key features of allergic airways disease at days 16 and 21 (figure 3C–J). In stark contrast, treatment of infected allergic groups did not reduce eosinophil or neutrophil recruitment to the airways. There were also no effects on H influenzae-induced IL-17, OVA-induced IL-5 or IL-13 release from MLN T-cell or AHR compared with untreated infected allergic groups at day 16 or day 21 (figure 3C–J). The exception was IFNγ, which was slightly but significantly increased by dexamethasone treatment on day 21 (figure 3H). To enhance clarity of the AHR data we have included a magnified online supplementary figure 1. To simplify the AHR data, we have shown dynamic compliance (figure 3K) and resistance (figure 3L) for all groups at one methacholine dose (5 mg/ml).
Collectively these data suggest that the induction of allergic airways disease promotes chronic infection, despite the presence of increased numbers of neutrophils, and leads to the development of neutrophilic allergic airways disease that is steroid resistant.
Chronic H influenzae infection in allergic airways disease does not affect MSC numbers or ciliary beat frequency
We then investigated the mechanisms involved in promoting chronic infection. Reduced mucus production and ciliary function in the airways may compromise bacterial clearance. Therefore, we assessed the effect of infection in allergic airways disease on MSC numbers and ciliary beat frequency in airway epithelia.
The induction of allergic airways disease significantly increased MSC numbers, but had no effect on ciliary beat frequency (figure 6A,B). Infection in allergic airways disease had no effect on either MSC numbers or cilia beat frequency compared with uninfected allergic controls (figure 6A,B).

Chronic Haemophilus influenzae (Hi) infection in allergic airways disease does not affect mucus-secreting cell (MSC) numbers or ciliary beat frequency. The effect of chronic H influenzae infection in allergic airways disease on (A) tissue MSCs and (B) ciliary beat frequency were assessed (day 16). Data represent the mean ± SEM from six to eight mice. Significant differences between allergic ovalbumin (OVA) and non-allergic (saline) groups are shown as ###p<0.001.
Chronic H influenzae infection in allergic airways disease inhibits innate immune cell activation and function
Innate immune responses are impaired in neutrophilic asthma. Therefore, we examined if chronic infection and neutrophilic allergic airways disease were associated with alterations in phagocyte function by assessing TLR expression and activation and function of neutrophils and macrophages from the lungs.
TLR2 and TLR4 expression is indicative of increased activation of neutrophils and macrophages. The induction of allergic airways disease increased the numbers of macrophages that were positive for TLR4 on days 16 and 21 but had no effect on the number of neutrophils expressing TLR2 or TLR4 or macrophages positive for TLR2 compared with uninfected allergic controls (figure 7A–D). Infection in allergic airways disease resulted in an increase in TLR2-positive macrophages but had no effect on TLR2 or four positive neutrophils or TLR4-positive macrophages on day 16. However, on day 21 the numbers of TLR4-positive neutrophils and macrophages were significantly reduced compared with allergic controls (figure 7A–D).

Chronic Haemophilus influenzae (Hi) infection in allergic airways disease inhibits innate immune cell activation and function. The effect of chronic Hi infection in allergic airways disease on neutrophil and macrophage (A–D) toll-like receptor 2 (TLR2) and TLR4 expression, and (E–F) activation status was examined by flow cytometry (representative flow cytometry plots of analysis of (A) TLR2 expression on neutrophils, (C) TLR4 and (E) CD62L on macrophages at day 16 are shown). The effects on (G) the numbers of phagocytosing neutrophils and macrophages, (H) oxidative burst and (I) serum antibody responses to Hi infection were assessed. Data represent the mean ± SEM from six to eight mice. Significant differences between allergic avalbumin (OVA) and non-allergic (saline (Sal)) groups are shown as ###p<0.001, ##p<0.01, #p<0.05. Significant differences between infected allergic (Hi /OVA) and uninfected allergic (OVA) groups are shown as **p<0.01, *p<0.05. Significant differences between infected allergic (Hi /OVA) and infected non-allergic (Hi) groups are shown as +p<0.05. Significant differences between infected allergic (Hi/OVA) and infected saline sensitised (Hi/Sal) groups are shown as ∨p<0.05. NS, non-significant.
Increased CD62L expression is a marker of reduced activation of innate immune cells. The induction of allergic airways disease had no effect on the number of CD62L-positive neutrophils and macrophages. However, infection in allergic airways disease resulted in a significant increase in CD62L-positive macrophages and neutrophils on day 16, indicating reduced activation compared with uninfected allergic controls (figure 7E,F).
We then investigated the function of neutrophils and macrophages in terms of phagocytosis and oxidative burst. The induction of allergic airways disease increased the numbers of phagocytosing neutrophils and macrophages in the lungs, but not oxidative burst in lung tissue. Critically, infection in allergic airways disease completely suppressed the numbers of phagocytosing neutrophils and macrophages in the airways (figure 7G) but increased oxidative burst compared with uninfected allergic groups on day 16 (figure 7H). Indeed the numbers of phagocytosing neutrophils and macrophages was reduced to the same levels as in uninfected non-allergic (saline) controls.
Allergic airways disease also reduced serum IgG1 responses to infection compared with saline sensitised groups (day 21, not day 16, figure 7I).
Chronic H influenzae infection and OVA sensitisation suppresses the numbers of phagocytosing macrophages
We then assessed whether the mechanisms that lead to chronic infection occur earlier in the timecourse, that is, with infection and OVA sensitisation, but prior to OVA challenge (day 11) to induce allergic airways disease (figure 8A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chronic Haemophilus influenzae (Hi) infection and ovalbumin (OVA) sensitisation suppress the numbers of phagocytosing macrophages. (A) Mice were infected, sensitised 10 days later, and infection and immune responses assessed on day 11. The effect of infection and sensitisation on (B) macrophage and neutrophil numbers in bronchoalveolar lavage fluid (BALF), (C) numbers of phagocytosing macrophages and neutrophils, and (D) antibody responses to Hi infection were assessed. Data represent the mean ± SEM from six to eight mice. Significant differences between infected allergic (Hi/OVA) and uninfected allergic (OVA) groups are shown as *p<0.05. Significant differences between infected allergic (Hi/OVA) and infected non-allergic (Hi) groups are shown as +p<0.05. Significant differences between infected allergic (Hi/OVA) and infected saline sensitised (Hi/Sal) groups are shown as ∨p<0.05.
Alveolar macrophage and neutrophil numbers in BALF were determined and there were no differences between groups (figure 8B). Macrophages were the predominant immune cell at this timepoint, however the number of phagocytosing macrophages was reduced in infected compared with uninfected OVA sensitised and infected non-sensitised controls (figure 8C). OVA sensitisation increased serum IgG1 responses to infection compared with saline sensitised groups (figure 8D), which may have been a result of the increased infection at this timepoint.
Collectively these results suggest that decreased activation and function of innate immune cells may contribute to the establishment of chronic infection in steroid-resistant neutrophilic allergic airways disease.
Discussion
We have recently showed that H influenzae drives the development of neutrophilic allergic airways disease that is dependent on IL-17.17 Here we extend these studies by showing that the combination of H influenzae infection and allergic airways disease results in chronic infection that drives the development of neutrophilic allergic airways disease that is steroid resistant. We also demonstrate that chronic infection in allergic airways disease impairs the activation and function of airway neutrophils and macrophages, which may promote bacterial persistence and airway neutrophilia. Notably the effects of infection were persistent and were evident before OVA challenge and for at least 5 days after the induction of allergic airways disease.
Chronic infection is commonly associated with many chronic airways diseases that are characterised by neutrophilic inflammation, such as COPD and bronchiectasis, and potentially neutrophilic asthma.4 ,7 ,15 ,16 ,24 Few studies have investigated the chronicity of infection in neutrophilic asthma, however an unexplained relationship between H influenzae and patients with neutrophilic asthma has been shown.4 ,15 This study contributes to our understanding of this relationship. A better understanding of the association may also identify the optimal targets for therapy (infection vs inflammation/immunity) that would benefit patients with neutrophilic asthma and those with other chronic airways diseases. A recent study showed that the airway secretions of 15% for all patients with asthma and 41% for patients with neutrophilic asthma had a significant load of potentially pathogenic bacteria, and that H influenzae was detected in 60% of the patients with a neutrophilic phenotype.15 Our data provide an understanding of this observation by demonstrating that the combination of an allergic environment and H influenzae infection results in the suppression of innate immune cell activation and function, which may lead to chronic infection. Interestingly, we show that allergic airways disease decreased antibody responses to infection compared with sham-sensitised groups, which may contribute to the persistence of infection.
Our results also show that a live H influenzae infection and the administration of killed H influenzae in allergic airways disease suppressed features of eosinophilic asthma, including eosinophilic inflammation and allergen-induced Th2 cytokines. Infection in allergic airways disease simultaneously induced features of neutrophilic asthma, including intense airway neutrophilic inflammation and AHR that was reduced compared with eosinophilic allergic airways disease but still elevated above non-allergic controls, as is the case in patients with neutrophilic asthma. Thus it is clear that exposure to bacteria suppresses the induction of allergic airways disease, which does not require the infectious process and may occur through altering sensitisation or challenge or both. Investigation of the mechanisms involved would require a detailed study of the effects of infection on different antigen-presenting cell, B-cell and T-cell populations, which is beyond the scope of this study. We have previously demonstrated that respiratory infection with Chlamydia also drives the development of neutrophilic allergic airways disease, and that the suppression of neutrophils during infection prevents the development of this phenotype.20
An important feature of patients with neutrophilic asthma is that they do not respond well to corticosteroids, which are the mainstay of asthma therapy.3 Our study demonstrates that all of the key features of infection-induced neutrophilic allergic airways disease, including airway eosinophils and neutrophils, Th1, Th2 and Th17 cytokine responses and AHR are resistant to dexamethasone treatment. Another study showed that severe combined immunodeficiency (SCID) mice reconstituted with Th17 cells were resistant to dexamethasone treatment, while treatment in mice reconstituted with Th2 cells significantly reduced inflammation and AHR. Importantly, in vitro, neutrophil apoptosis is resistant to steroid action, demonstrating that steroids prolong neutrophil survival.25 Several molecular mechanisms have been suggested to explain steroid resistance, such as increases in glucocorticoid receptor (GR)-β which binds to DNA and not steroids.3 Interestingly, IL-17 is able to induce the expression of GR-β mRNA in asthmatic epithelial cells in vitro.26 Taken together, this study and others suggest that infection-induced increases in neutrophilic inflammation, Th17 cells and IL-17 production may be important in promoting steroid resistance in patients with neutrophilic asthma.
Steroid treatment caused a small reduction in bacterial numbers in allergic airways disease at day 16. Treatment substantially exacerbated infection in non-allergic groups after the infection had apparently cleared. This suggests that the infection persisted at undetectable levels, perhaps intracellularly, and could be reactivated by steroid treatment, which is known to suppress immune responses.3
We also investigated the mechanisms involved in the induction of chronic infection. Chronic infection was maintained in allergic airways disease despite the increase in airway neutrophils in infected allergic groups compared with uninfected allergic controls. In the healthy airway, normal mucociliary clearance of inhaled particles and bacteria requires the interaction of normally beating respiratory cilia and the overlying mucus blanket. Reduction of ciliary function and mucus production in the airways may compromise bacterial clearance. Infection and inflammatory cells have been reported to impair ciliary beat frequency and mucociliary clearance.22 However, we found that there were no reductions in MSCs or ciliary beat frequency in infected allergic compared with uninfected allergic groups, suggesting that alterations in these innate immune defences are not responsible for chronic infection in neutrophilic asthma.
The most important cellular components of innate immunity are neutrophils and macrophages, which when activated mount an immediate non-specific response and clear bacteria by phagocytosis. We therefore investigated the combined effects of infection and allergic airways disease on the activation and function of airway neutrophils and macrophages. TLRs are expressed on neutrophils and macrophages and increases in their activation and signalling are involved in the activation, chemokine receptor expression and function of these phagocytic cells.27 Although TLR2 is important, TLR4 is crucial for host defence and clearance of pulmonary H influenzae infection.28 We show here that in infected allergic groups, TLR2-positive cells were unchanged compared with uninfected allergic controls (at days 16 and 21), the only exception being an increase in TLR2-positive macrophages on day 16. Interestingly, we show that, on day 21, TLR4-positive neutrophils and macrophages were reduced compared with allergic controls, demonstrating that H influenzae in allergic airways disease suppresses the influx of TLR4-positive innate immune cells, which may contribute to the persistence of infection.
Neutrophil and macrophage activation was also investigated. Activation of neutrophils and macrophages occurs in stages and includes a range of phenotypic and functional changes. Low levels of activation lead to the modulation of surface receptors and priming to subsequent stimuli, while higher levels enhance function.29 ,30 CD62L (L-selectin) is a surface receptor that is highly expressed on resting neutrophils and circulating monocytes, and mediates their migration to the site of inflammation. During normal activation, migration and maturation, CD62L expression is downregulated by shedding and its expression is indicative of reduced activation and maturity.30–32 Our study demonstrates that chronic H influenzae infection during allergic airways disease results in increased numbers of neutrophils and macrophages expressing CD62L. This indicates that H influenzae may suppress the activation of these cells, which may contribute to chronic infection in neutrophilic asthma.
The function of neutrophils and macrophages was also examined. Once activated, neutrophils and macrophages gain microbicidal functions, the most important of which is phagocytosis and elimination of invading pathogens.33 ,34 Critically we show that infection in allergic airways disease completely inhibited the influx and/or development of phagocytosing neutrophils and macrophages in the airways compared with uninfected allergic controls. The mechanisms that underpin this inhibition of phagocytosis in infected allergic groups are currently unknown and are under investigation. However, possible mechanisms include increased apoptosis of phagocytosing neutrophils and macrophages, increased necrosis of these cells, or altered pathogen recognition receptor signalling. There were significantly less phagocytosing neutrophils and macrophages in infected allergic compared with uninfected allergic groups at days 11, 16 and 21. There were also fewer of these phagocytosing cells in infected allergic compared with infected alone groups but only at days 11 and 16 and not at day 21. These results suggest that it may be the asthma phenotype, rather than the effects of infection, that plays the dominant role in driving reduced phagocytosis. However, infection may also contribute to the decreased function of phagocytes.
The lack of activation and influx of phagocytes may be the reasons for the development of chronic infection in neutrophilic allergic airways disease and in neutrophilic asthma. However, this does not rule out the possibility that other mechanisms of innate immune dysfunction may be responsible for chronic infection and neutrophilic asthma. There is the potential that the increased neutrophilic inflammation observed in neutrophilic asthma may be the result of the increase in neutrophil influx to compensate for their decreased function.
Our results also show that lung cells from infected allergic groups generated more oxidative burst than uninfected allergic controls. Oxidative burst increases oxidative stress and has significant effects on the pathophysiology of asthma, such as increasing inflammation and airway smooth muscle contractility.35 Increased oxidative stress occurs in patients with asthma who have increased bacterial load compared with healthy controls, which is probably due to the bacterial load rather than increased neutrophilic inflammation.15
Early in the model, we show increased antibody responses to H influenzae infection in OVA-sensitised groups, which may be a result of increased infection. However, later, the combination of infection and allergic airways disease reduced antibody responses and this could also contribute to chronic infection. The reduction in antibody responses presumably results from a suppression of B-cell activity that requires T-cell help. We show similar suppressive effects on T-cell activity.
Taken together these results may explain the association of infection with neutrophilic asthma. Allergic airways disease promotes persistent infection and induces the influx of defective innate immune cells that fail to clear the bacteria but may increase oxidative stress.
In conclusion, we show that the combination of allergic airways disease and H influenzae infection leads to chronic infection. Chronic infection together with infection-induced increases in Th17 cells and IL-17 production may lead to the development of neutrophilic allergic airways disease and asthma. We demonstrate that this phenotype is steroid resistant, which may be the result of a combination of increased neutrophils and Th17 cells. Chronic infection probably results from the suppression of innate cell activation and function, however further studies of the mechanisms of neutrophilic asthma are required to better understand disease progression. Such studies would enable the development of more effective treatments for patients with this disease phenotype, whereby targeting the infection and asthmatic responses may be beneficial.
References
Footnotes
Funding Hunter Medical Research Institute, the National Health and Medical Research Council of Australia, The Australian Research Council, and University of Newcastle.
Competing interests None.
Ethics approval All experiments were approved by the Animal Care and Ethics Committee, University of Newcastle.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves