Article Text

Abstract
Chronic obstructive pulmonary disease (COPD) is a common and important disease. Neutrophils have been shown to play a fundamental role in its development and progression. Understanding the mechanisms underlying the trafficking of neutrophils across the vascular endothelium into the lung could potentially allow the development of targeted biological treatments. The early stages of neutrophil tethering, adherence to and rolling on the endothelium have been determined. The later stages of diapedesis through the glycocalyx, endothelial cell (EC) layer and basement membrane, which are less well characterised, have been reviewed here. Evidence obtained from in vitro and in vivo work, concerning the implicated adhesion molecules on the neutrophil and endothelium, the mechanisms for neutrophil navigation through the EC junction (paracellular route) and evidence for transmigration through the body of an EC itself (transcellular route), is considered. The mechanisms are complex and are often disease and stimulus specific. There is evidence that a significant degree of redundancy occurs. Transmigration in the lung differs from that in other organs in that the neutrophil can exit the circulation either through the postcapillary venule in the systemic circulation or through the capillary in the pulmonary circulation. A number of factors make the mechanisms of transmigration within the lung and COPD model unique. These include physical differences between the flow through the capillary and the postcapillary venule, the modulating effect of the alveolar epithelium and other cells such as the macrophage, the presence of a ‘diseased’ neutrophil and indeed the presence or absence of acute, acute on chronic or chronic pulmonary disease.
- COPD mechanisms
- neutrophil biology
- cytokine biology
- COPD pathology
- emphysema
- asthma
- asthma mechanisms
- COPD exacerbations
- cough/mechanisms/pharmacology
- airway epithelium
- allergic lung disease
- asthma mechanisms
- cytokine biology
- eosinophil biology
- innate immunity
- lymphocyte biology
Statistics from Altmetric.com
- COPD mechanisms
- neutrophil biology
- cytokine biology
- COPD pathology
- emphysema
- asthma
- asthma mechanisms
- COPD exacerbations
- cough/mechanisms/pharmacology
- airway epithelium
- allergic lung disease
- asthma mechanisms
- cytokine biology
- eosinophil biology
- innate immunity
- lymphocyte biology
Introduction
Chronic obstructive pulmonary disease (COPD) is a common and disabling illness. It is projected to become the third most common cause of death worldwide by 2020.1 In addition to a fundamental role in lung defence against pathogens, neutrophils play a critical role in driving COPD.2 Understanding how neutrophils are recruited from the circulation to the airway may help gain insight into why some, but not all smokers develop COPD and may potentially assist in guiding the development of targeted immune-modulating treatments.
Trafficking of leucocytes out of the circulation into tissues is a highly regulated, complex, multistep process. The sequence of events begins with margination of the cell to the wall of the vessel, tethering to and then rolling of the leucocyte on the vascular endothelium, followed by tight adhesion to the endothelium.3 The leucocyte then migrates laterally to an appropriate point to transmigrate (diapedese) across the vessel.4 Neutrophil transendothelial migration (TEM) occurs within minutes in a stimulated endothelium.5 6 The cell surface adhesion molecules involved in the initial steps of rolling, tethering and adhesion have been well characterised.3 The mechanisms by which the neutrophil then migrates through the vessel wall have not yet been fully determined. This article will review the evidence regarding these steps and then focus more specifically on neutrophil TEM in the lung and COPD model.
Structure of the vascular endothelium and its cell–cell junctions
The normal vascular endothelium consists of an endothelial cell (EC) layer, with a basement membrane (BM) below and glycocalyx on the luminal side (figure 1). The glycocalyx consists of proteoglycans and glycolipids, along with other functional proteins on its surface.7 The BM contains collagen IV, laminins, nidogens and perlecan.8–10 Embedded in the BM and outside it is a non-continuous layer of cells known as pericytes.8 These are found on the outer surface of capillaries as well as other blood vessels and may have a role in a number of vascular processes such as angiogenesis.11 12 Vascular endothelium can be studied in vivo (in animals), or can be cultured in vitro, most commonly from human umbilical vein endothelial cells (HUVECs).
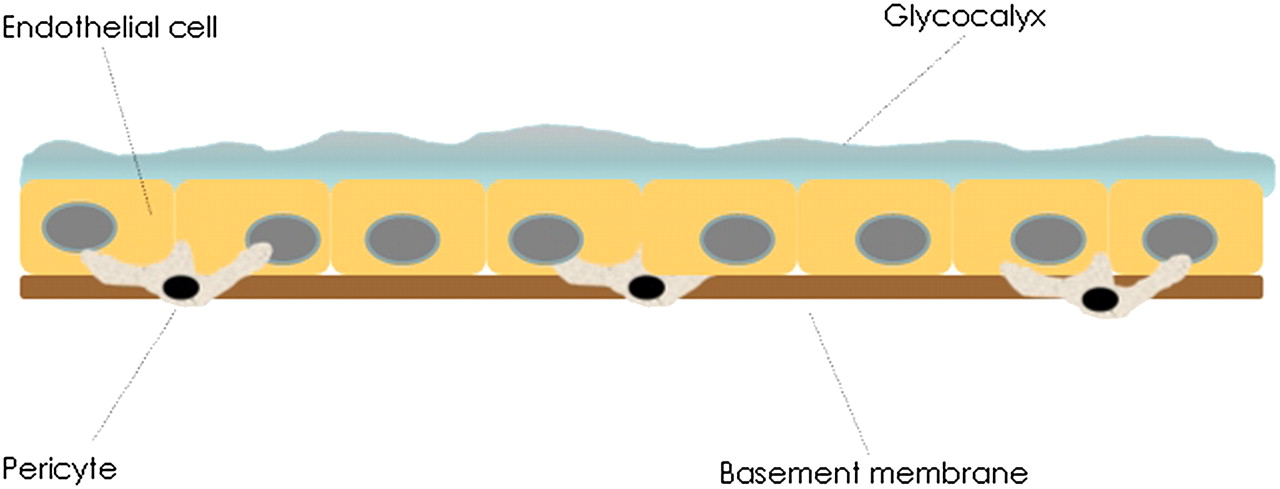
The vascular endothelium. The neutrophil has to navigate three components of the endothelium to reach the extracellular matrix: the glycocalyx, endothelial cell layer and the basement membrane containing pericytes embedded within it.
The components of the vascular endothelium have to be traversed by the emigrating neutrophil. Each EC is adhered to its adjacent cells. It is the ability of these cells to allow gap formation for passage of fluid, proteins and leucocytes that is a key role of the endothelium, but one which must be tightly regulated. A number of cell–cell junction molecules have been studied and reviewed.13 14 These are outlined in table 1.13–15 Junctional molecules bind homophilically to allow junctional adhesion to occur.
The principal endothelial intercellular junctional complexes
It is well recognised that significant differences exist between ECs found in different vascular beds in vivo, with clear functional advantages.16 For example, an increased number of tight junctions are found between ECs in the brain in order to maintain the blood–brain barrier13 and fenestrations between ECs are abundant in the kidney to permit filtration. Expression of cell surface adhesion molecules also varies between sites.16 These site-specific variations must be borne in mind when considering the results obtained from in vitro and in vivo animal work.
Paracellular transmigration
Transmigration via an EC junction usually occurs at the intersection of three or more ECs, known as the tricellular corner.17 18 Numerous studies have been undertaken to determine the adhesion molecules involved in moving the neutrophil from the apical surface of the endothelium through to the extracellular matrix. Figure 2 gives an illustrative summary of the paracellular and transcellular routes.

Overview of the paracellular and transcellular routes of transmigration. (A) Neutrophil extending a dominant pseudopod to invaginate the apical endothelial cell (EC) membrane. Inset: endothelial adhesion molecules, in this case, ICAM-1 clustering under the migrating neutrophil and binding to lymphocyte function-associated antigen 1 (LFA-1). PECAM-1 has also been seen to cluster. Movement through the EC may potentially be facilitated by formation of a pore through fusion of submembranous vesicles and/or by proteases.4 19 20 21–25 (B) Neutrophil migrating between two ECs. (C) Inset: a polarised neutrophil seen laterally migrating to the EC junction. ICAM-1 again clusters beneath the neutrophil. The likely positions of the EC adhesion molecules are illustrated. Not all the molecules will be involved in every case of neutrophil transendothelial migration.20 26–30 31 A ‘low expression region’ in the basement membrane is also seen and this corresponds to the EC junction and a gap between pericytes.8 ESAM, endothelial cell selective adhesion molecule; ICAM, intercellular adhesion molecule; JAM, junctional adhesion molecule; PECAM, platelet endothelial cell adhesion molecule.
A number of key principles have been learnt from these experiments. First, while it is possible for some neutrophils to migrate across an unstimulated EC monolayer in vitro, this process is greatly enhanced by stimulation of the EC and/or the neutrophil (leading to upregulation of adhesion molecules) and by the presence of a chemotactic gradient across the endothelium.19 32–39 Stimulants include cytokines, such as tumour necrosis factor α (TNFα) or interleukin 1β (IL-1β), endogenous endothelial-bound neutrophil chemoattractants including the chemokines leukotriene B4 (LTB4), C5a or IL-8, or exogenous stimulants such as the bacterial endotoxin, lipopolysaccharide.5 34 36–40 The same principle also applies in vivo.
A number of molecules involved in neutrophil TEM have been identified by applying monoclonal antibodies (mAbs) to block known cell surface molecules on ECs in vitro/in vivo and/or to the neutrophils used in the experiments. Most of the studies have been able to separate the role of specific molecules on neutrophil adherence from a role in the later processes of TEM. The most studied of these transmembrane proteins belong to two families, the β2-integrins (CD18/CD11) and the immunoglobulin superfamily. The principal molecules involved in TEM and their ligands are shown in table 2. Stimulation of the EC or the neutrophil generally, but not always, leads to upregulation of these molecules.19 34 35 45
The adhesion molecules that have been identified as playing a role in neutrophil TEM
The EC adhesion molecules involved specifically in TEM have been shown to localise to certain points on the apical surface of the cell or within the EC junction (illustrated in figure 2).
The mechanisms of TEM are clearly complex. Studies have shown that different combinations of adhesion molecules mediate TEM, dependent in part on the stimulus employed.32 34 Woodfin et al injected mice with either IL-1β or TNFα and then exteriorised and examined the cremaster muscle venules using intravital microscopy, a technique which allows real-time imaging of the microcirculation. Experiments involved mice with double knockout of specific adhesion molecules or mAbs administered to wild-type mice. The two cytokines were shown to act in different ways to allow neutrophil TEM. IL-1β-stimulated TEM was mediated by intercellular adhesion molecule-2 (ICAM-2), junctional adhesion molecule-A (JAM-A) and platelet endothelial cell adhesion molecule-1 (PECAM-1) on the endothelium. This was shown by reduced TEM in the presence of the relevant blocking mAbs to these adhesion proteins, whereas transmigration was not affected by the presence of blocking antibodies to ICAM-2, JAM-A and PECAM-1 in a TNFα-stimulated model. Further experiments using mAbs demonstrated that TNFα stimulates TEM at least in part via macrophage antigen-1 (Mac-1) (on neutrophils) and JAM-C (on ECs).26
The authors also demonstrated the principle that the same cytokine can promote TEM in different ways. Experiments using TNFα receptor-negative mice or wild-type mice, injected with either wild-type neutrophils or TNFα receptor-negative neutrophils, demonstrated that when stimulated with TNFα, specific activation of the endothelium is essential for both neutrophil adhesion to the endothelium and TEM to occur. Activation of the neutrophil in the TNFα-stimulated mouse is not required for adhesion but is necessary to allow full neutrophil TEM.
In addition, the key adhesion molecules exhibit the phenomenon of redundancy. A number of authors have shown that in the experimental model at least, blocking one or more adhesion molecules can cause compensatory TEM involving other adhesion molecules.32 34 37
The adhesion molecule itself may have an influence on the degree of TEM. Sachs et al demonstrated that a greater number of CD177-positive neutrophils transmigrated across a HUVEC monolayer in comparison with CD177-negative neutrophils.35
These and other studies indicate that there remains some way to go before the exact sequences of neutrophil TEM are fully elucidated, especially when one considers the different organ systems and the likely modulating influence of different disease processes.
While studies employing mAbs have clearly shown the involvement of β2-integrins and other molecules, they do not identify the mechanism by which neutrophils are actually able to transmigrate across the endothelium.
Neutrophil navigation through an EC junction
A neutrophil which has arrested and adhered to the endothelium undergoes polarisation or shape change and then migrates laterally until a suitable point for diapedesis has been found (in general an EC junction). Crawling may even occur in the opposite direction to blood flow.4 The mechanisms behind these processes are beyond the scope of this review, but in brief the neutrophil changes shape in order that it has a leading edge and a uropod distally and produces cellular processes (pseudopods) that can probe the endothelial membrane.46 47 Ortolan et al showed using a HUVEC model and an mAb that inhibition of CD157 on the neutrophil or EC led to longer migration distances by the neutrophil before TEM. This suggests that CD157 plays a role in signalling to the neutrophil to diapedese at a suitable point.28 In mouse models, Mac-1 and its ligand ICAM-1 play a fundamental role in the crawling process.4 Yang et al also demonstrated that at 24 h poststimulation with TNFα, HUVECs had changed from a polygonal shape to a more elongated form, with the potential advantage of reducing the distance required for neutrophils to crawl to reach the nearest EC junction for diapedesis.19
Other EC adhesion molecules have been shown to relocate, for example ICAM-1 clusters beneath adherent neutrophils.19 It may be that these molecules contribute to endothelial ‘docking stations’ seen with electron microscopy.21 Woodfin et al demonstrated that ICAM-2, JAM-A and PECAM-1 act sequentially in their interactions with neutrophil ligands at different points in the EC junction (figure 2).26
The main junctional complexes are outlined in table 1. There is conflicting evidence as to how these junctional proteins are dealt with by the neutrophil. Studies using electron microscopy have shown that tight junctions are present as circumferential binding rings at the apices of adjacent ECs, with an absence of staining at tricellular corners.17 The majority of neutrophils were shown to migrate around tight junctions, most frequently but not always at tricellular corners.17 48 Staining for adherens junction was seen to be discontinuous along borders, particularly at tricellular corners, whereas PECAM-1 (which binds homophilically on adjacent ECs) was continuous.17 Su et al demonstrated in cultured HUVECs and in an ex vivo model using actual umbilical vein endothelium, that VE-cadherin appears to move back and away on each side of a transmigrating neutrophil (without being disrupted), whereas PECAM-1–PECAM-1 complexes open sideways around the neutrophil in a zip-like effect. Both junctional complexes are seen to return to their original positions after TEM has occurred.18 This may explain why a proportion of neutrophils are able to diapedese between two adjacent ECs in addition to the tricellular corner where junctional complex proteins are sparser. Other studies have suggested that neutrophil proteases are responsible for degrading junctional proteins in order for the neutrophil to diapedese.15 49 50 This conflicts with work suggesting that TEM can occur independently of proteases.45
It is likely that, despite methodological differences and technical limitations between the studies, multiple mechanisms for leucocyte TEM exist.
The EC itself plays a role in creating a physical gap for the neutrophil to be able to squeeze through. Garcia et al showed that adherence of neutrophils to the endothelium led to an increase in activation of EC intracellular myosin light chain kinase, responsible for phosphorylation of myosin light chains. This leads to cytoskeleton contraction and subsequent intercellular gap formation.51 Rabodzey et al suggested that the force of the neutrophil cellular processes known as pseudopodia,19 which probe and then move into the intercellular junction (probably as a result of EC–neutrophil interactions) are able to ‘force apart’ adherens junctions.52
The effect of the shear force of blood flow on the endothelium is not always recreated in in vitro experiments. Although its presence does affect the degree of leucocyte TEM in vitro,19 others have argued that the presence of a glycocalyx in vivo will attenuate the effect of shear force.53
Two studies examining the role of JAM-C have produced conflicting results.20 27 This again illustrates the challenges of interpreting these experiments, as in these examples both the endothelial stimulant and presence of shear flow differed. Burns et al attempted to reproduce the transendothelial forces that are present as plasma itself moves across the endothelium under inflammatory conditions. The authors concluded that nitric oxide produced by the EC in response to this force could inhibit TEM.54
Neutrophil–EC ligand binding may occur purely in some instances to guide the neutrophil through the EC junction. O'Brien et al demonstrated by transfecting ECs with genetically altered PECAM-1 molecules that PECAM-1 probably plays a passive neutrophil binding role in TEM.39 It is certain, however, that binding of some molecules triggers intracellular signalling with consequent downstream effects on TEM.22 55
Diapedesis through the BM and pericyte layer
It is important that leucocytes traverse the BM without causing extensive damage, in order to preserve its role in barrier integrity and structural support to the vessel.10 Wang et al demonstrated in an in vivo mouse model using immunofluorescent staining and confocal microscopy regions within the BM in which there is a reduced expression of the key matrix proteins laminins and collagen IV. These were termed ‘low expression regions’ (LERs) and were shown to align with EC junctions and gaps between pericytes.8 9
The majority of neutrophils were observed to be adherent to the endothelium in close proximity to an underlying LER and gap between pericytes. The same group also demonstrated that the majority of neutrophils were seen to migrate between the EC layer and BM to LERs.9 Staining of fixed tissue showed that neutrophils appeared to pass directly through the LER.8 In vitro studies have also demonstrated subendothelial migration,56 prior to BM transmigration.
Different leucocytes possess different mechanisms to pass through the LER, in the same way that they can do at other stages of TEM.8 19 32 56 Monocytes were shown to change their morphology to a greater extent than neutrophils in order to ‘squeeze’ through the LER. Neutrophils on the other hand were able to increase the size of the LER.8 9 The presence of laminin on the surface of 20% of transmigrated neutrophils and the inhibition of transmigration by blocking antibodies to neutrophil elastase in this and other studies suggests that elastase may play a role in the size increase seen in LERs.8 57 As with other stages of TEM, the redundancy phenomenon exists and compensatory mechanisms are in place (as neutrophil elastase-deficient mice have no defect in TEM).8 57
Neutrophil β1- and β3-integrins have been identified as ligands for BM matrix proteins.56–58 Neutrophil–EC interaction via proteins such as PECAM-1 during transmigration through the EC layer are likely to play a role in upregulation of the neutrophil adhesion molecules required for BM transmigration.57 Kenne et al have shown that matrix proteins in the BM are not passive molecules to be traversed but play an active role, as laminin α-4 knockout mice demonstrated impaired neutrophil diapedesis.59
While these studies suggest that neutrophils bypass the pericytes through gaps between these cells, other work has suggested (at least under specific inflammatory experimental conditions) that the majority of neutrophils appear to traverse both the EC and the pericyte in a transcellular manner.60
Transcellular transmigration
Transcellular migration is an extensive topic in its own right and involves the diapedesis of leucocytes through the body of the EC, bypassing the EC junction (figure 2). The evidence for transcellular diapedesis in all leucocyte classes, obtained initially from in vivo work and, more recently, in vitro studies, has been extensively reviewed elsewhere, but will be discussed in brief here with respect to neutrophils.23
In the past, limitations of both light microscopy and single-section 2D electron microscopy have made it difficult to determine (with confidence) that diapedesing leucocytes which appear to be passing through the body of an EC, are in fact not just migrating via an EC junction which has not been imaged in the chosen plane. However, recent advances to technology have allowed more detailed reconstruction of images to overcome this potential problem. Phillipson et al used confocal microscopy to produce stacked images taken at progressive levels through the endothelial wall in their genetically altered mouse model.
By producing these images and layering them against images of the same vessel stained for EC junctions with fluorescent-labelled anti-PECAM-1, they demonstrated that neutrophils in Mac-1-deficient mice had delayed TEM (in response to macrophage inflammatory protein 2 (MIP-2)) and were more likely to use a transcellular route (61% compared with 14% of wild-type neutrophils).4 These appearances correlated with 2D electron microscopy images.4 21
The interesting questions of why and how leucocytes undergo transcellular diapedesis remain incompletely answered. The work by Phillipson et al (among others) demonstrated that Mac-1-deficient neutrophils were unable to crawl along the endothelium and were seen to develop pseudopods which probed the EC membrane.4 21 60 These experimental conditions suggest it is possible that leucocytes possess the ability to undergo transcellular transmigration to a greater extent when the paracellular route is unavailable or more difficult to utilise. A study in guinea pigs60 and another using an in vitro model22 demonstrated that the majority of neutrophils utilised the transcellular route, which is in contrast to other studies.15 19 21 24 It may be that the proportion of leucocytes transmigrating via the transcellular rather than the paracellular route is stimulus and site specific and/or dependent on the experimental model or leucocyte being studied.
As with all these studies, variations in experimental conditions between studies make reaching definitive conclusions difficult. Luu et al showed that the percentage of neutrophils diapedesing across an endothelial monolayer (specific route not assessed) was reduced when certain cytokines were presented to the HUVECs during flow across the surface of the EC layer, to simulate systemic inflammation, as opposed to by direct stimulation of the HUVECs.5
The adhesion molecules involved in transcellular neutrophil TEM are becoming characterised. Yang et al have studied transcellular migration in a HUVEC model and shown that after 24 h of stimulation with TNFα, a small but significant proportion of neutrophils used a transcellular route.19 This corresponded to an increased expression of endothelial ICAM-1 at this time point compared with 4 h, but was unrelated to the number of neutrophils adhering to the EC. Live-cell epifluorescence imaging showed these neutrophils passing through the ECs at points slightly away from junctions, which were stained with non-blocking VE-cadherin antibodies. To lend further support, ICAM-1 was observed to actively cluster under the neutrophil, in the same way that it does beneath transmigrating leucocytes using the paracellular route.25 Transfection of a HUVEC line with an ICAM-1 construct led to enhanced EC expression of the protein and increased transcellular migration at both 4 and 24 h. Blocking antibody studies to lymphocyte function-associated antigen 1 (LFA-1) and Mac-1 suggested that LFA-1 is the more relevant ligand for ICAM-1 in the context of transcellular migration. Both routes were enhanced by but were not dependent on the presence of shear flow. Other authors have shown transcellular diapedesis occurring in HUVEC layers, using other stimulants.15 22 24
Yang et al demonstrated that neutrophils developed cellular processes, similar to pseudopods, in contact with and then probing through a ‘hole’ in the EC membrane.19 Exactly how these processes might be involved in allowing the neutrophil to pass through the body of the EC remains unclear. Phillipson et al published electron micrographs showing discontinuations within the basal EC membrane adjacent to a diapedesing neutrophil.21 Whether or not passage through the cell and its membrane is due to proteases or pore formation or another mechanism requires further study. Carmen et al have demonstrated in in vitro lymphocyte work that fluorescently labelled ICAM-1 and PECAM-1 were seen to form multiple ring-like structures under the crawling lymphocyte, followed by formation of pores in the EC. Pore formation was dependent on probing by a lymphocyte cellular process into the EC membrane and the presence of increased numbers of subendothelial membrane vesiculo-vacuolar organelles which may fuse to allow creation of a pore.24 Interestingly, Phillipson et al also suggested from their images that the endothelial membrane changes shape to produce a ‘docking station’ for the neutrophil (both wild type and Mac-1 deficient), which in some images was seen to progress to a dome-like structure covering the leucocyte and preventing increased endothelial permeability to proteins.21 Williams et al showed that cleaved annexin 1, a secretory product of neutrophils, was required in order for ICAM-1 to cluster and form docking stations. An mAb to annexin 1 reduced transcellular TEM, providing supporting evidence for this mechanism.22 Van Buul et al also demonstrated the presence of endothelial docking stations (termed ‘cup-like protusions’ by the authors) partially surrounding the adherent leucocyte (in this case a neutrophil-like leucocyte from a promyelocytic cell line).61 Formation of this ‘cup’ was dependent on binding of the leucocyte to EC ICAM-1, leading to recruitment and activation of the endothelial small GTPase RhoG, with subsequent actin cytoskeleton rearrangement. Interestingly, depletion of the endothelial RhoG decreased leucocyte TEM by >70%. The authors hypothesised therefore that given the degree of TEM inhibition, endothelial cup formation may also be necessary for paracellular transmigration. However, the route of transmigration was not specifically assessed and it may therefore be, as the authors discuss, that RhoG plays more than one role in TEM.
The lung vasculature
The lung derives its blood supply from both the pulmonary and bronchial arteries.62 Blood from the former circulation passes into the alveolar capillary bed to allow gas exchange and the latter supplies nutrients and oxygen to the bronchi, vessels and lung parenchyma.63 Significant anastomosis occurs between the bronchial and pulmonary circulation.64
Figure 3 illustrates the relationship of the (bronchial) capillary bed within the airway wall and its relationship to the bronchial epithelium.64 The circulatory supply to the alveolus (figure 4) is different in that on one side of the alveolar wall the EC layer of the capillary lies in close apposition to the alveolar epithelial cell layer, with a shared BM. The other side of the alveolus has a thicker wall due to extracellular matrix produced by fibroblasts.7 65

The circulatory supply to the bronchus. Simplified cross-sectional representation of a medium-sized bronchus. The area highlighted by a circle demonstrates the positions of the interconnected subepithelial and submucosal capillary networks, which are derived from the systemic bronchial circulation. Neutrophils entering the airway lumen from the bronchial circulation do so via the postcapillary venule.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The circulatory supply to the alveolus. Neutrophils in the pulmonary circulation transmigrate across the endothelium from the capillary bed rather than the postcapillary venule.
TEM in the lung and COPD model
The lung is unique in that TEM occurs not only through the postcapillary venules of the systemic circulation but also via the alveolar capillaries arising from the pulmonary circulation.66 The diameter of the capillaries is often less than that of the neutrophil, leading to slower leucocyte transit time through the vessel67 68 and obviating the need for rolling on the endothelium.69 While L-selectin is therefore not required for classical rolling, Doyle et al demonstrated in a mouse model that its presence on the endothelium was still required under certain inflammatory stimuli to retain neutrophils for longer in the capillary segment.69
Despite these structural features, it has been argued that significant neutrophil sequestration within the lung occurs only under certain circumstances, for example following neutrophil activation prior to entry into the pulmonary circulation.70
Much of the research concerning TEM in the lung has been conducted in models designed to simulate acute pulmonary infection or acute lung injury secondary to sepsis at a distant site. Neutrophils are more likely to sequester in the lung capillaries following an inflammatory stimulus.69 71 72 In work by Drost et al, neutrophils showed upregulation of Mac-1 following cytokine stimulus, suggesting the neutrophil was then primed for TEM.71 Reduced deformability of the neutrophil also occurs and has been confirmed in real-life human sepsis.72
Neutrophil TEM in the lung can also occur via a β2-integrin-independent mechanism.32 Interestingly, this may depend on whether the background inflammatory stimulus is acute or chronic.36 Mackarel et al harvested neutrophils from stable or exacerbating patients with COPD, cystic fibrosis or bronchiectasis and compared these with healthy neutrophils. The healthy neutrophils and those from stable patients transmigrated across a human pulmonary artery endothelial cell (HPAEC) monolayer using a CD18 (β2-integrin)-independent pathway in response to IL-8 or LTB4, whereas neutrophils from acutely exacerbating patients migrated in a CD18-dependent manner. The stimulus-specific nature of this process is seen in that neutrophil TEM in response to fMLP (N-formyl-methionyl-leucyl-phenylalanine) was CD18 dependent in the acute, chronic and normal groups.36 In a mouse pneumonia model, ICAM-1 was identified as the ligand for CD11/CD18 integrins.32 Further studies are clearly needed to determine the specific role of other adhesion molecules in neutrophil TEM in the lung in the presence of disease states.
Walker et al demonstrated the presence of small ‘holes’ within the basal laminae of the alveolar capillary in rabbits and mice with pneumonia, which neutrophils were ‘squeezing’ through.73 In a rat model, shear forces were shown to be reduced in the capillaries in comparison with the postcapillary venules. Thus the effect of shear forces on TEM across the capillary may be less than in the postcapillary venule.74 Walker et al showed that neutrophils can utilise the transcellular route in pulmonary venules.65
The inherent complexity of neutrophil TEM in the lung/COPD model is illustrated further by several other observations. First, the capillary endothelium does not act in isolation. Weppler et al developed an in vitro bilayer model consisting of an endothelial and alveolar epithelial cell layer. They demonstrated in two studies that the epithelial component can exert an inhibitory effect on neutrophil TEM (depending again upon the inflammatory stimulus employed).75 76 In vivo experiments illustrate the role that other inflammatory cells play in coordinating neutrophil TEM. CCR2 is a key monocyte-attractant chemokine receptor. A study using CCR2 knockout mice showed impaired neutrophil and monocyte TEM. Further experiments confirmed this and suggest that under specific stimuli monocytes and neutrophils may undergo ‘cross-talk’, with one in part facilitating the other's transmigration at different time points.40 ‘Cross-talk’ also occurs between cell surface molecules/receptors within the same cell77 and applies to other inflammatory cell types such as the eosinophil.78
Alveolar macrophages also play a role. In a mouse peritonitis model, production of platelet-activating factor and hydrogen peroxide by alveolar macrophagess led to endothelial superoxide production. Blockade of superoxide caused a subsequent reduction in neutrophil TEM.79 In addition, in sepsis secondary to peritonitis, the chemokine concentration will be greater in the circulation than in the lung and yet neutrophils are able to transmigrate, apparently against this gradient.
Chemokines/cytokines are clearly important in attracting inflammatory cells to the lung in COPD. The interplay between mediators produced by activated epithelial cells and macrophages and subsequent neutrophil, monocyte and T cell influx is complex and has been reviewed elsewhere.80 CXCL8 (IL-8), CXCL1 (Gro-α) and CXCL5 (ENA-78) are key neutrophil chemoattractants, secreted by macrophages, which activate the receptors CXCR1 (IL-8) and CXCR2 (IL-8, Gro-α and ENA-78).80 Antagonising these receptors or blocking the intracellular enzymes involved in coordinating production of cytokines/chemokines (such as phosphodiesterase-4) may provide an alternative to the development of drugs which directly block endothelial transmigration itself.80 81
Lastly, the ‘diseased’ neutrophil itself may influence the process of transmigration. A number of studies have attempted to determine if neutrophils from patients with COPD have any specific defining characteristics. Several studies have found a greater expression of the β2-integrin Mac-1.82–85 In one study, Mac-1 persisted for a longer period during the apoptotic process in stable patients with COPD compared with controls.86 Others have not found Mac-1 upregulation.87 Woolhouse et al demonstrated that COPD neutrophils transmigrated in greater numbers across a flow-stimulated HUVEC layer in comparison with those from healthy smokers, non-smokers and, interestingly, patients with COPD due to α1-antitrypsin deficiency.88 Conversely, in Mackarel's study,36 neutrophils from patients with chronic pulmonary diseases transmigrated to a lesser extent in response to some stimuli than those from normal subjects. In this latter study a HPAEC layer was used. Sapey et al recently demonstrated that neutrophils from patients with stable COPD undergo chemotaxis with greater speed but less accuracy in response to several stimuli, compared with neutrophils from healthy smokers or matched α1-antitrypsin-deficient patients with COPD.89 These findings raise the question as to whether these differences in neutrophil function are seen as a consequence of established ongoing disease or whether in fact the possession of a subset of neutrophils with variant function predisposes individuals to develop a disease.
Limitations, complexities and future research needs
The in vitro and in vivo studies discussed above have produced compelling evidence for the complexity of mechanisms involved in neutrophil TEM in general. There are, however, limitations to the available techniques that are of importance. While in vitro work allows the use of human cells, it is difficult to know to what extent this accurately reflects processes occurring in vivo. HUVECs, which originate from large vessel endothelium, may differ functionally from the ECs found within different vascular beds. Standard HUVEC culture techniques do not produce a glycocalyx or BM, which in vivo is likely to exert effects on TEM.53 59 90 Some authors have attempted to use more organ-specific monolayers, for example human lung microvascular endothelial cells or HPAECs.24 36 Accurately reproducing a disease state (or even ‘normal’ endothelium) in an in vitro model is also difficult due to the complex interactions of the cells and molecular messengers involved. For this reason alone, in vivo work has obvious advantages, but again there are likely to be significant differences between humans and other animals. Investigators have attempted to produce animal models of COPD using a variety of methods such as cigarette smoke. Unfortunately as yet these models are unable to reproduce reliably the complex heterogeneous condition that is COPD. Cigarette smoke can induce an emphysema-like response in the mouse, but producing a model with chronic bronchitis, pulmonary hypertension or small airways disease has not yet been achieved.63
It would be important to study some of the above findings within a COPD model. For example, are neutrophils from patients with COPD less deformable than those of controls? Does this vary with exacerbations? What are the effects of exacerbations on TEM mechanisms? Do mechanisms vary with stage and phenotype of disease? Do patients with COPD possess a subset of neutrophils that can undergo reverse transendothelial migration (as has been seen in other inflammatory conditions)91 and how could this relate to the systemic effects of the disease?
While these and many other questions remain to be answered, there has been considerable progress in recent years in determining the later steps of neutrophil migration from the circulation into target tissues. The complexity and redundancy of the involved mechanisms makes developing biological treatments to modulate neutrophil TEM a significant challenge, unless specific and/or universal defects of the process can be identified.
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.