Article Text

Abstract
Rationale Bacterial pneumonia is the most common infectious cause of death worldwide and treatment is increasingly hampered by antibiotic resistance. Mesenchymal stem cells (MSCs) have been demonstrated to provide protection against acute inflammatory lung injury; however, their potential therapeutic role in the setting of bacterial pneumonia has not been well studied.
Objective This study focused on testing the therapeutic and mechanistic effects of MSCs in a mouse model of Gram-negative pneumonia.
Methods and results Syngeneic MSCs from wild-type mice were isolated and administered via the intratracheal route to mice 4 h after the mice were infected with Escherichia coli. 3T3 fibroblasts and phosphate-buffered saline (PBS) were used as controls for all in vivo experiments. Survival, lung injury, bacterial counts and indices of inflammation were measured in each treatment group. Treatment with wild-type MSCs improved 48 h survival (MSC, 55%; 3T3, 8%; PBS, 0%; p<0.05 for MSC vs 3T3 and PBS groups) and lung injury compared with control mice. In addition, wild-type MSCs enhanced bacterial clearance from the alveolar space as early as 4 h after administration, an effect that was not observed with the other treatment groups. The antibacterial effect with MSCs was due, in part, to their upregulation of the antibacterial protein lipocalin 2.
Conclusions Treatment with MSCs enhanced survival and bacterial clearance in a mouse model of Gram-negative pneumonia. The bacterial clearance effect was due, in part, to the upregulation of lipocalin 2 production by MSCs.
- Mesenchymal stem cells
- macrophages
- lipocalin 2
- pneumonia
- innate immunity
- ARDS
- respiratory infection
Statistics from Altmetric.com
Key messages
What is the key question?
What are the therapeutic and mechanistic effects of mouse bone marrow derived mesenchymal stem cells (MSCs) administered via the intratracheal route in a murine model of Gram-negative pneumonia?
What is the bottom line?
MSCs significantly reduce mortality and lung injury and enhance bacterial clearance from the lung when delivered after the establishment of Escherichia coli pneumonia in mice.
Why read on?
The antibacterial effect observed with MSC treatment is explained, in part, by their capacity to upregulate expression and secretion of lipocalin 2, a known antimicrobial factor, in response to inflammatory stimuli.
Introduction
Bacterial pneumonia is the third most common cause of death worldwide, the most common cause of death in developing countries, and the most common infectious cause of death worldwide.1 The incidence of antibiotic resistance among bacterial isolates has been increasing at a rapid rate, thereby greatly limiting the armamentarium of medications available to treat patients with bacterial pneumonia.2 Therefore, given the large public health impact of bacterial pneumonia and the decreasing supply of effective antimicrobials, novel therapies are needed.
The innate immune response against bacterial pathogens in the lung classically consists of resident alveolar macrophages, recruited neutrophils, and endogenous antimicrobial factors present in the respiratory secretions. There is increasing evidence that lipocalin 2, which inhibits bacterial growth by blocking iron uptake, is one of the critical proteins involved in the innate immune response to bacterial infection in the lung. It has been recently demonstrated that lipocalin 2 is needed for effective host defence against pulmonary infection by Escherichia coli,3 Mycobacterium tuberculosis4 and Klebsiella pneumoniae.5 The lack of lipocalin 2 expression resulted in increased morbidity and mortality of infected mice, particularly in models of Gram-negative pneumonia.3 5 Although lipocalin 2 has been shown to be produced by multiple cell types, including neutrophils, macrophages and epithelial cells, there has also been recent literature reporting that a stromal cell line from bone marrow (ST2) may be a source of lipocalin 2.6
Mesenchymal stem cells (MSCs) are multipotent, adult stem cells that are classically isolated from the bone marrow. Recent literature has demonstrated that these cells have potent immunomodulatory properties7–9 including the suppression of pro-inflammatory processes in the lung in response to endotoxin and bleomycin.10–15 Given the predominantly immunosuppressive phenotype of MSCs, there has been concern over their effect on host defence to a live bacterial infection. Since bacterial pneumonia and sepsis are the two most common causes of respiratory failure from acute lung injury and acute respiratory distress syndrome,16 understanding the effect of MSCs in these settings is important. A recent study by Nemeth et al17 demonstrated that MSCs improved survival in a cecal ligation and puncture model of sepsis and led to lower bacterial counts in the blood. However, the mechanism by which MSCs enhanced bacterial clearance was not presented. Also, a recent publication by Mei et al demonstrated that MSCs can enhance the phagocytic capacity of host immune cells when administered in a mouse model of sepsis.18 Another recent study by our group19 provided evidence that human MSCs directly release the antibacterial peptide LL-37 in response to bacterial infection. We conducted this study to investigate the therapeutic and mechanistic effects of murine MSCs in a well characterised model of bacterial pneumonia in mice20 21 and to specifically determine the role of lipocalin 2 in the antimicrobial effect of MSCs in this model.
Methods
Please see online supplement for more details.
Animal care
C57BL/6 male mice (8–10 weeks old; Jackson Laboratory) were used in all experiments. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco and the University of Pittsburgh.
Experimental design
The experimental design was similar to that used by our group previously,10 with the exception that lung injury was induced with the intratracheal instillation of live E coli K1 bacteria at a concentration of 106 cfu/25 μl of phosphate-buffered saline (PBS).
Cell cultures
Wild-type mouse MSCs were isolated and expanded from the bone marrow of green fluorescent protein positive (GFP+)-C57BL/6 mice using the same protocol described previously with the exception that cells were used at passage numbers 5–10 not at higher passages as used in the previous study.10 Lower passage numbers were used in this study because recent literature has demonstrated that long-term cultures of MSCs are predisposed to malignant transformation and loss of biological activity.22 23 Cells fulfilled the criteria for MSCs as established by the International Society of Cellular Therapy.24
Protein assays
Cytokines and lipocalin 2 levels were measured using standard ELISA kits from R&D (Minneapolis, MN, USA). Myeloperoxidase was measured using standard methods25 as a way to quantify neutrophil degranulation.
In vivo lipocalin 2 blocking studies
In vivo lipocalin 2 blocking studies were performed to determine the importance of the upregulation of lipocalin 2 observed with MSC treatment, and its relationship to the antibacterial effect.
Lipocalin 2 production by lipopolysaccharide-stimulated MSCs
Conditioned media from unstimulated and lipopolysaccharide (LPS)-stimulated MSCs were obtained, and lipocalin 2 was measured using an ELISA kit (R&D).
Alveolar macrophage isolation and stimulation
Alveolar macrophages were isolated from normal C57BL/6 mice by bronchoalveolar lavage (BAL) using calcium- and magnesium-free PBS supplemented with 0.6 mM ethylenediaminetetraacetic acid (EDTA). The cells were then plated and stimulated with either LPS (±MSCs in a transwell) or with conditioned media obtained from LPS-stimulated MSCs. Supernatants were collected and analysed by ELISA for lipocalin 2 and tumour necrosis factor α (TNFα) levels.
Stimulation of MSCs with alveolar macrophage conditioned media and inflammatory cytokines
MSCs were stimulated with either conditioned media from LPS-stimulated alveolar macrophages or with inflammatory cytokines in the presence of LPS. The supernatant from these studies was used to measure lipocalin 2 concentrations.
Statistical analysis
Comparisons between the two groups were made using an unpaired t test. The log-rank test was used for comparing survival data. A value of p<0.05 was considered statistically significant. Analyses were done using SPSS software and GraphPad Prism software. Data are shown as mean±SD.
Results
MSCs improve survival and phenotype in the E coli pneumonia model
MSC-treated mice had a significantly higher rate of survival over 48 h compared with fibroblast (3T3)-treated and PBS-treated mice (figure 1). MSC treatment also resulted in an improvement in the activity and phenotype of the mice at 48 h compared with fibroblast-treated and PBS-treated mice.
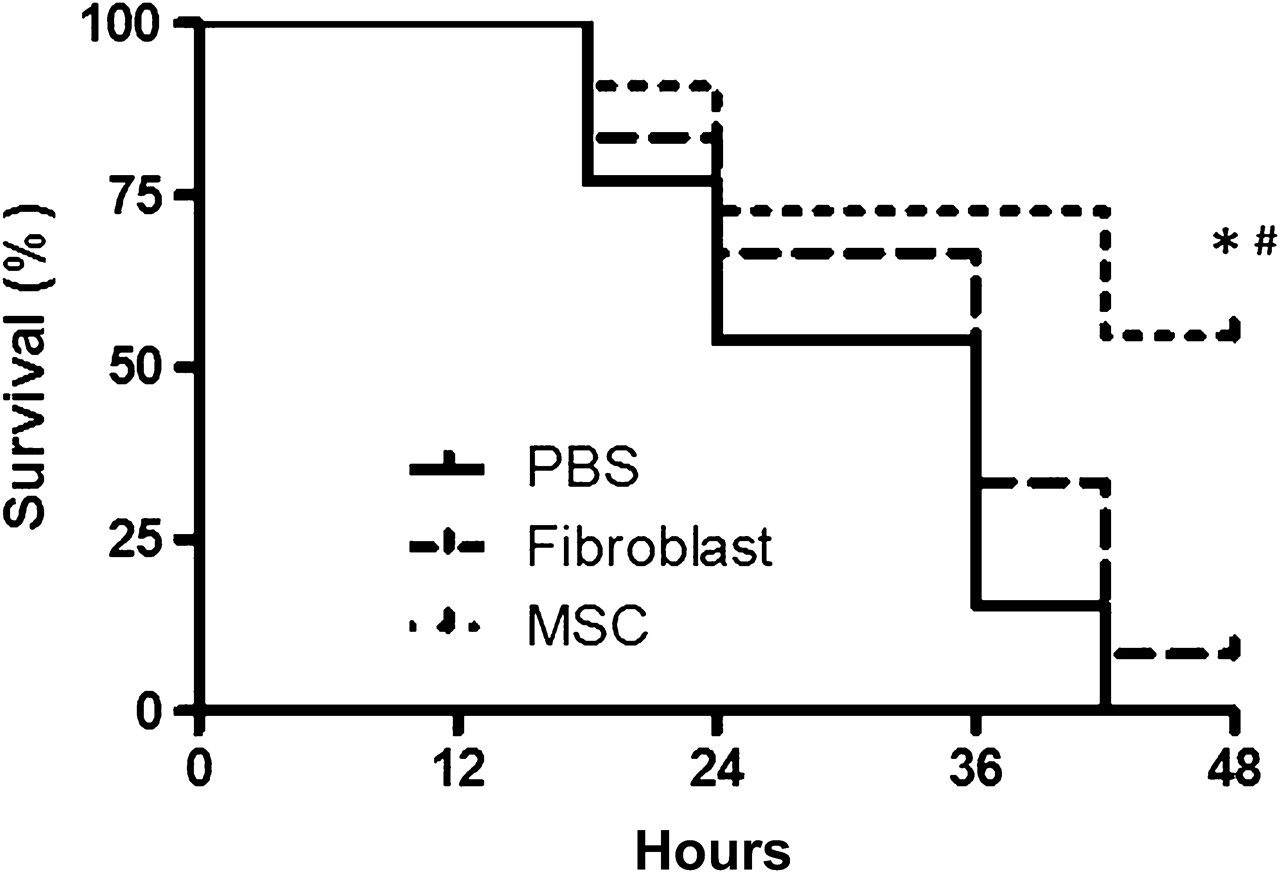
Intratracheal mesenchymal stem cell (MSC) treatment improved survival in the Escherichia coli pneumonia model of acute lung injury. Mice were injured with 106 cfu of E coli instilled intratracheally and then were given either MSCs, 3T3 fibroblasts (750 000 MSCs or 3T3 fibroblasts/30 μl phosphate-buffered saline (PBS)) or PBS (30 μl) as treatment 4 h later. Survival over 48 h was determined in each group, which was significantly higher in the MSC-treated mice (n=11–12 per group, *p<0.01 for MSC vs PBS and #p=0.03 for MSC vs 3T3 group, using a log-rank test).
MSCs reduce the severity of lung injury in the E coli pneumonia model
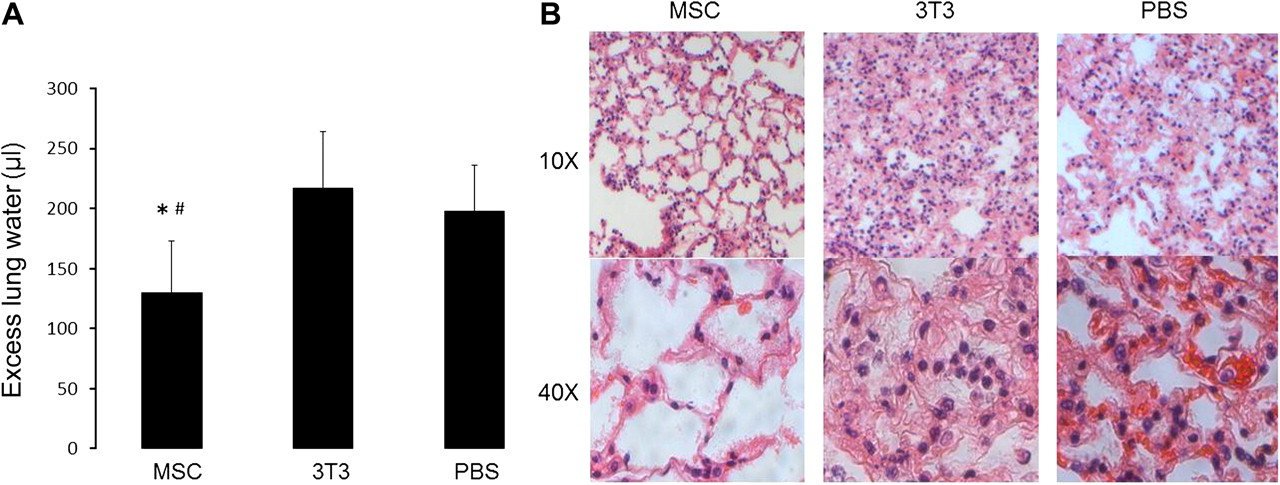
MSC-treated mice had a significant reduction in the severity of lung injury as measured by excess lung water (figure 2A). The quantity of excess lung water was measured for mice in each group at the time of death or at the endpoint of the experiment (48 h). Histological analysis also demonstrated qualitatively less lung injury in the MSC-treated mice compared with the other groups (figure 2B). Histology was obtained from a mouse from each group that survived the longest in order to accurately compare the severity of lung injury between groups. Immunohistochemistry for GFP expression demonstrated no detectable GFP+ MSCs in the lung at 48 h after infection with E coli.

Mesenchymal stem cell (MSC) treatment reduced the severity of lung injury in Escherichia coli pneumonia. (A) Mice treated with MSCs had a lower quantity of excess lung water compared with mice treated with 3T3 fibroblasts and phosphate-buffered saline (PBS) (n=14–15 per group, *, #p<0.0001 for MSC vs PBS and MSC vs 3T3 group, respectively). Data are mean±SD. (B) Haematoxylin and eosin staining of lung tissue demonstrated that MSC-treated mice had qualitatively less lung injury than mice treated with 3T3 fibroblasts or PBS.
MSC treatment enhanced bacterial clearance from the lung as early as 4 h after instillation
Treatment with MSCs enhanced bacterial clearance from the alveolar space as early as 4 h after instillation. This was determined by culturing the BAL fluid 4 h after treatment with MSCs or control (figure 3A).

Mesenchymal stem cell (MSC) treatment led to enhanced bacterial clearance after infection with intratracheal Escherichia coli. (A) Mice treated with MSCs had fewer E coli cfu in their bronchoalveolar lavage (BAL) at 8 h post infection compared with control-treated mice (n=12 per group, *p=0.002 for MSC vs phosphate-buffered saline (PBS) and #p=0.04 for MSC vs 3T3 group). Data are mean±SD. (B) MSC treatment also led to a reduction in the number of E coli cfu in the whole lung homogenate 24 h after infection (n=8–9 per group, *p=0.01). Data are mean±SD.
This reduction in bacterial burden persisted at 24 h when the number of E coli in the whole lung homogenate was measured in MSC-treated and control mice (figure 3B).
MSCs reduced the early pro-inflammatory response to Ecoli pneumonia
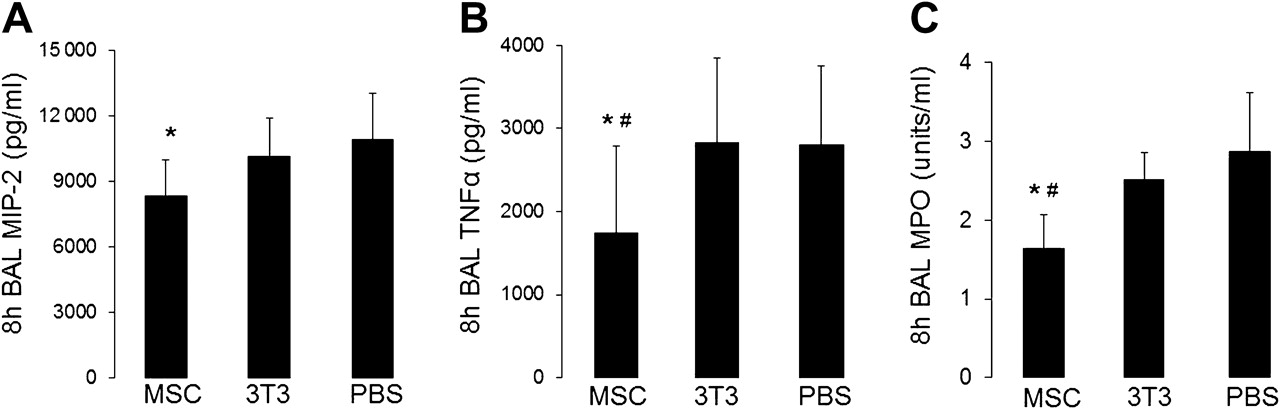
Treatment with MSCs reduced the early pro-inflammatory response to E coli instillation as measured by 8 h BAL levels of macrophage inflammatory protein 2 (MIP-2) and TNFα (figure 4A,B). MSC-treated mice also had evidence of less neutrophil degranulation in the alveolar space as assessed by BAL levels of myeloperoxidase (figure 4C). MSC treatment had no effect on the level of interleukin 10 (IL-10) in the BAL fluid compared with control mice (data not shown).

Mesenchymal stem cell (MSC) treatment reduced the early pro-inflammatory response to Escherichia coli pneumonia. (A) Macrophage inflammatory protein 2 (MIP-2) was significantly lower in MSC-treated mice versus phosphate-buffered saline (PBS)-treated mice (n=7–8 per group, *p=0.03) and (B) tumour necrosis factor α (TNFα) was reduced in MSC-treated mice compared with 3T3-treated and PBS-treated mice 8 h after infection (n=8–10 per group, *, #p=0.04 for MSC vs PBS and 3T3 groups). Data are mean±SD. (C) Neutrophil influx was reduced in MSC-treated mice compared with controls as measured by bronchoalveolar lavage (BAL) levels of myeloperoxidase (MPO) 8 h after infection with E coli (n=4 per group, *p=0.04 for MSC vs PBS and #p=0.03 for MSC vs 3T3). Data are mean±SD.
MSCs upregulate the concentration of lipocalin 2 in the alveolar space
Levels of lipocalin 2 were quantified by ELISA, which demonstrated a 56% increase in the quantity of lipocalin 2 in the BAL fluid of MSC-treated mice compared with PBS-treated mice (figure 5A). Other antimicrobial factors were also screened for, including defensins and collectins such as surfactant protein-D, but the results were no different for MSC-treated and control mice.
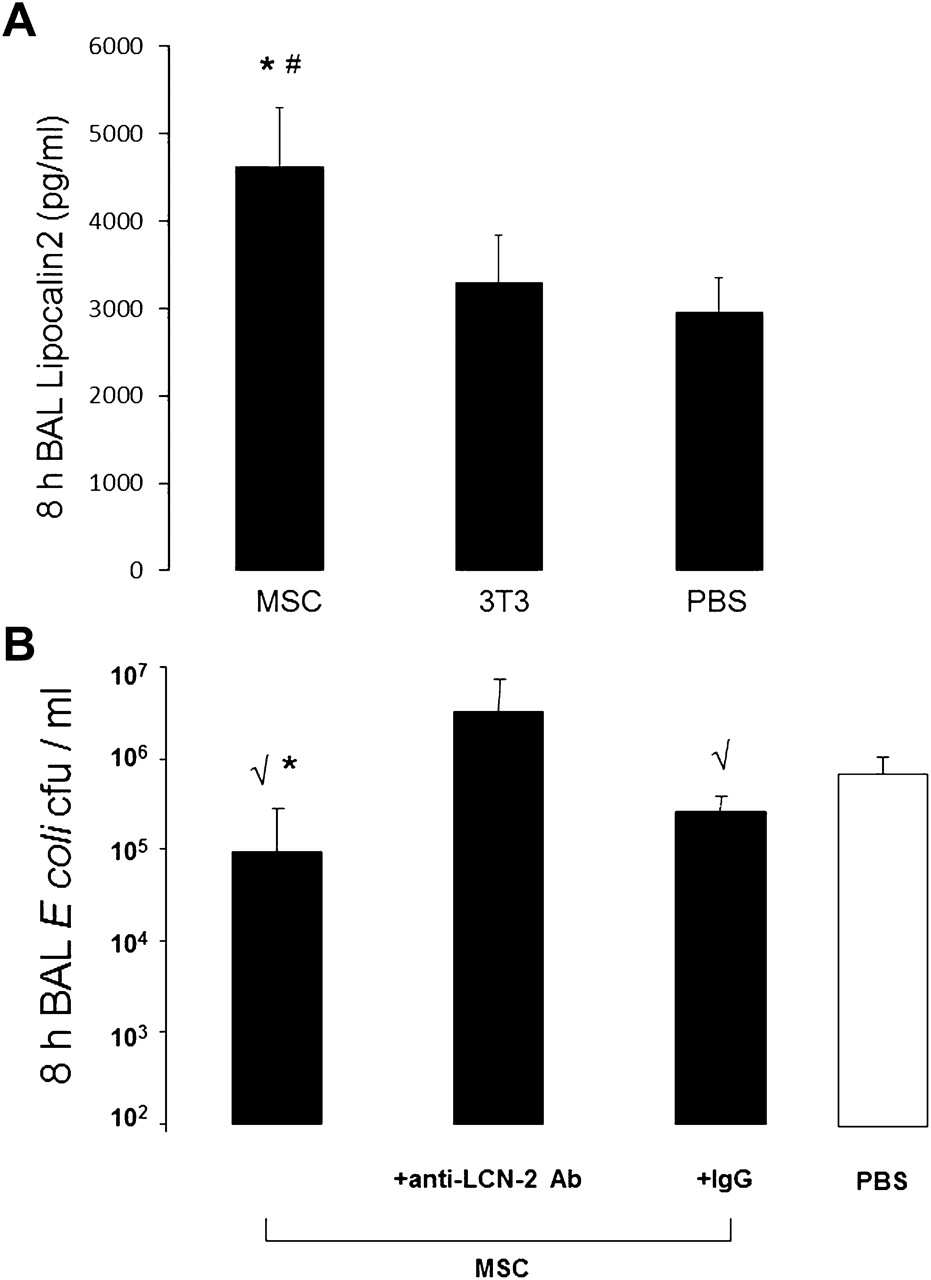
Lipocalin 2 production is upregulated with mesenchymal stem cell (MSC) treatment and accounts for part of the antimicrobial effect. (A) MSC-treated mice had higher levels of lipocalin 2 in their bronchoalveolar lavage (BAL) fluid at 8 h post infection (n=4–6 per group, *p=0.005 for MSC vs phosphate-buffered solution (PBS) and #p=0.01 for MSC vs 3T3). Data are mean±SD. (B) Co-administration of a lipocalin 2 blocking antibody (anti-lcn-2 Ab) with MSCs significantly reduced the antibacterial effect of MSC treatment in vivo. MSCs retained significant antibacterial activity when co-administered with an isotype control antibody (n=3–7 per group, √p<0.01 vs PBS; *p=0.035 vs anti-lcn-2 Ab). Data are mean±SD.
Blocking lipocalin 2 inhibits the in vivo antibacterial activity of MSCs
To determine if lipocalin 2 was an important mediator of the bacterial clearance effect seen with MSC treatment, blocking studies were done using the in vivo model of E. coli pneumonia. When the lipocalin 2 blocking antibody was premixed with MSCs at the time of cell delivery, the antibacterial effect of MSC treatment was eliminated compared with control mice. When MSCs were premixed with the isotype control antibody, the cells had preserved antibacterial activity compared with PBS-treated mice (figure 5B).
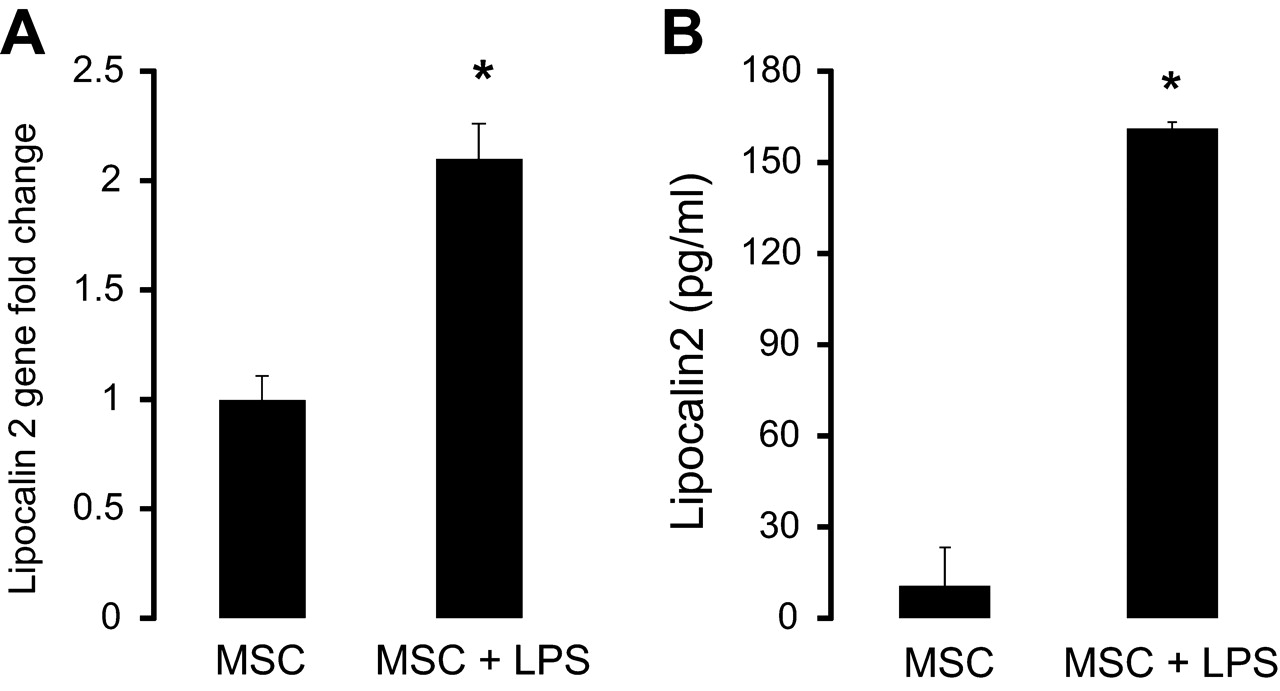
MSCs upregulate mRNA and protein production of lipocalin 2 in response to LPS stimulation
To determine whether MSCs, themselves, were an important source of lipocalin 2 or other known antimicrobial proteins, quantitative reverse transcriptase PCR was carried out for lipocalin 2 gene expression in unstimulated and LPS-stimulated MSCs. As seen in figure 6A, MSCs increased their transcription of the lipocalin 2 gene by approximately twofold upon LPS stimulation.

Mesenchymal stem cells (MSCs) upregulate their gene expression and secretion of lipocalin 2 with lipopolysaccharide (LPS) stimulation. (A) Quantitative reverse transcriptase PCR on RNA isolated from unstimulated and LPS-stimulated MSCs demonstrated a significant upregulation of mRNA for the lipocalin 2 gene with LPS stimulation (n=5 per group, *p<0.00001 for MSC + LPS vs MSC). Data are mean±SD. (B) MSCs upregulate secretion of lipocalin 2 after stimulation with LPS (n=4 per group, *p<0.000001 for MSC + LPS vs MSC). Data are mean±SD.
To determine if MSCs produced lipocalin 2 protein in a measureable quantity, cells were stimulated with LPS for 24 h to allow for adequate time for protein synthesis and secretion and then the conditioned media was collected to measure lipocalin 2 levels by ELISA. As shown in figure 6B, MSCs significantly upregulated production of lipocalin 2 protein with LPS stimulation, which is consistent with the enhanced mRNA expression observed. However, the absolute quantity of lipocalin 2 in the MSC-conditioned media was still relatively low compared with that measured in the BAL fluid from in vivo experiments (figure 5A).
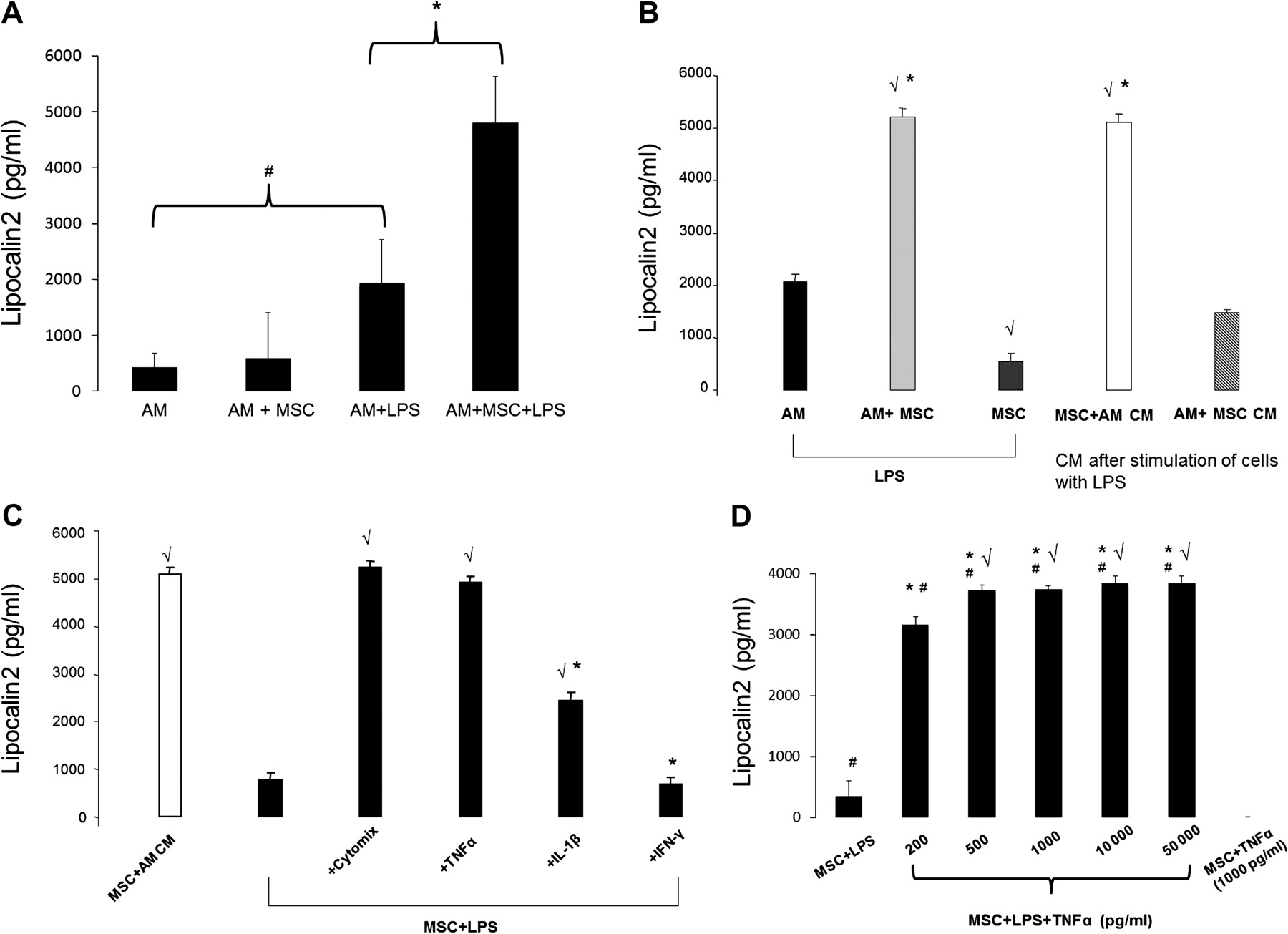
Co-culture of LPS-stimulated MSCs and alveolar macrophages enhances production of lipocalin 2
Given that the amount of lipocalin 2 produced by MSCs was much less than that measured in the BAL fluid obtained from in vivo experiments, we hypothesised that MSCs upregulated lipocalin 2 production by innate immune cells in the lung based on previous literature demonstrating that neutrophils and macrophages can contribute to intrinsic lipocalin 2 production in the lung.4 26 The principal immune cell present in the lung during the initial inflammatory response to bacterial infection is the alveolar macrophage. Because MSCs were delivered by the intratracheal route in this study, we hypothesised that the interaction between MSCs and alveolar macrophages may contribute to enhanced lipocalin 2 production.
To test this hypothesis, MSCs were co-cultured with alveolar macrophages in a transwell system and stimulated with LPS for up to 24 h. At this time, the media was collected and lipocalin 2 measured by ELISA. As shown in figure 7A, culturing MSCs and alveolar macrophages together in the presence of LPS resulted in a synergistic effect on lipocalin 2 production compared with levels measured by either cell type alone. The presence of the transwell to physically separate the MSCs and macrophages suggested that the upregulation of lipocalin 2 production occurred by a paracrine mechanism. Furthermore, the amount of lipocalin 2 detected under these conditions was comparable to that measured in the in vivo BAL samples, suggesting that the interaction between MSCs and alveolar macrophages was relevant to the in vivo findings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mesenchymal stem cells (MSCs) enhance their secretion of lipocalin 2 in response to a combination of inflammatory signals including tumour necrosis factor α (TNFα), produced by activated macrophages, and lipopolysaccharide (LPS). (A) Co-culture of MSCs and alveolar macrophages (AMs) led to a significant increase in the quantity of lipocalin 2 produced (n=8–12 per group, *p<0.0000001, #p<0.0001). (B) The conditioned media of LPS-stimulated AMs augmented MSC secretion of lipocalin 2, whereas the conditioned media of LPS-stimulated MSCs had no effect on AM production of lipocalin 2 (n=4 per group, √p<0.00001 vs AM + LPS group, *p<0.0000001 vs MSC + LPS group). (C) Incubation of MSCs with cytomix resulted in an increase in lipocalin 2 production commensurate with that seen with LPS-stimulated AM-conditioned media. This effect was driven primarily by TNFα (n=3–4 per group, √p<0.0001 vs MSC + LPS group, *p<0.001 vs MSC + LPS + TNFα group). (D) Incubation of LPS-stimulated MSCs with TNFα resulted in a significant increase in lipocalin 2 production starting at 200 pg/ml of TNFα; in the absence of LPS, TNFα had no effect on lipocalin 2 production by MSCs (n=4 per group, *p<10−11 vs MSC + LPS group, √p=0.0003 vs MSC + LPS + 200 pg/ml TNF-α group, #p<0.05 vs MSC + 1000 pg/ml TNFα). Data are mean±SD.
Conditioned media from LPS-stimulated alveolar macrophages enhances lipocalin 2 production by MSCs, an effect that is recapitulated by TNFα
To test our original hypothesis that MSCs enhanced alveolar macrophage production of lipocalin 2, we incubated alveolar macrophages with conditioned media obtained from LPS-stimulated MSCs and measured the amount of lipocalin 2 that was produced. However, contrary to our hypothesis, the addition of LPS-stimulated MSC conditioned media did not result in an increase in lipocalin 2 levels compared with stimulation of alveolar macrophages with LPS alone (figure 7B). Unexpectedly, incubation of MSCs with LPS-stimulated alveolar macrophage conditioned media led to a pronounced increase in lipocalin 2 production, similar to that observed when both cells were incubated together in the presence of LPS (figure 7B).
Given that alveolar macrophages are well known to produce significant quantities of inflammatory mediators in response to endotoxin or bacterial stimulation, we hypothesised that the principal mediators involved in upregulating MSC production of lipocalin 2 could be the pro-inflammatory cytokines TNFα and IL-1β. This hypothesis was also derived from recent studies that reported that IL-1β and TNFα have the capacity to upregulate cellular production of lipocalin 2 at the transcriptional level.27–30 Therefore, in the presence of LPS, MSCs were incubated with TNFα, IL-1β, and interferon γ (IFNγ) simultaneously (cytomix) and individually to assess their effect on MSC production of lipocalin 2. As shown in figure 7C, cytomix led to an upregulation of lipocalin 2 production by MSCs in a quantity similar to that seen with LPS-stimulated alveolar macrophage conditioned media. The strongest effect on lipocalin 2 production by MSCs was seen with TNFα, while there was no effect with IFNγ.
Therefore, the amount of TNFα was measured in LPS-stimulated alveolar macrophages (3080±642 pg/ml), and in the presence of LPS, MSCs were then incubated with a range of TNFα concentrations to reflect the quantity produced by alveolar macrophages in vitro and what was measured in vivo (figure 4B). As shown in figure 7D, TNFα stimulated MSC production of lipocalin 2 at a concentration as low as 200 pg/ml and resulted in maximal production at 500 pg/ml, suggesting that physiological levels of TNFα are sufficient to induce upregulation of lipocalin 2 in MSCs. However, in the absence of LPS, TNFα had no effect on lipocalin 2 production by MSCs, indicating that LPS is a required cofactor in the stimulatory effect.
Discussion
There are three main findings of the current study: (1) MSCs reduce lung injury and improve survival in a model of Gram-negative bacterial pneumonia; (2) MSCs enhance bacterial clearance in the alveolar space as early as 4 h after administration, while still retaining their classic immunosuppressive properties; (3) MSCs significantly upregulate their production of lipocalin 2 in response to LPS and inflammatory mediators generated by activated macrophages, and this response contributes to the antibacterial effect observed with MSC treatment.
The finding that MSC treatment can lead to enhanced bacterial clearance from the lung is of particular importance because MSCs have been thought to have a primarily immunosuppressive phenotype and therefore a potentially deleterious effect on host defence to bacterial infection. To explain the antibacterial effect observed with MSC treatment, we hypothesised that lipocalin 2, a protein that binds bacterial siderophores and restricts iron utilisation by bacteria, may be an important antimicrobial factor. Our data demonstrated that treatment with MSCs did lead to a significant upregulation of lipocalin 2 levels in the alveolar space, and that blocking lipocalin 2 in vivo eliminated the bacterial clearance effect observed with MSCs. These results suggest that lipocalin 2 is an important mediator of the antibacterial effect seen with MSC treatment in the model of pneumonia used in this study. It is important to note that the general role of lipocalin 2 in host defence to pulmonary infection is unclear as there are reports of the ability of clinical isolates of the Gram-negative bacteria K pneumoniae to cause respiratory infection through evasion of the inhibitory effects of lipocalin 2.31
Following the finding that lipocalin 2 is an important antibacterial protein upregulated by MSC administration, we focused on determining the cellular sources of lipocalin 2 in this model. We carried out a series of in vitro experiments that demonstrated that alveolar macrophages stimulated MSCs to enhance their secretion of lipocalin 2 to levels comparable to that measured in vivo. This effect was recapitulated by TNFα stimulation in the presence of LPS. It is important to note that the ability of TNFα to upregulate lipocalin 2 production by MSCs was observed in the presence of concurrent LPS stimulation, which was added to the conditions to model the effect of LPS-stimulated alveolar macrophage conditioned media. When MSCs were stimulated with TNFα alone there was no increase in lipocalin 2 production. These data are consistent with previous studies that have shown that TNFα alone does not lead to upregulation of lipocalin 2.27 30 TNFα requires a costimulus through either the IL-17 receptor pathway or the IL-1 receptor/toll-like receptor pathway to activate transcription of the lipocalin 2 gene.27 30 The finding that MSCs can upregulate its production of the antimicrobial protein, lipocalin 2, in response to infectious and inflammatory signals, such as LPS and TNFα, is important since it demonstrates the ability of MSCs to sense the local environment and respond in a manner to help restore homeostasis in the host. Furthermore, this result provides evidence that MSCs can function as a member of the innate immune system to infection, and suggests that MSCs might be used therapeutically to bolster the innate immune response to bacterial infection.
There are still some limitations and unanswered questions from this study. Despite the finding that MSCs can produce significant quantities of lipocalin 2 in response to stimulation with LPS and TNFα, we do not know the contributions to lipocalin 2 production by other cell types in the lung and other innate immune cells. It is well known that neutrophils and lung epithelial cells make lipocalin 2,4 5 26 27 so understanding more completely the relative contributions from other cell types will be important. This can likely only be assessed through a combination of strategies using selective deletion of cell populations and genetically modified mice deficient for lipocalin 2.
Also, additional work will be needed to determine if camp, the mouse homologue of the antibacterial peptide LL-37, interacts with the production of lipocalin 2. Our group recently published a study demonstrating that human MSCs secrete LL-37 in quantities sufficient to kill bacteria in a similar Gram-negative pneumonia model.19 However, it is unclear what the relevance of this finding will be in mouse MSCs, which is the focus of the current study. Therefore, it will be necessary to determine if mouse MSCs secrete camp, and if this results in direct bacterial killing or participates in the regulation of the secretion of other antimicrobial proteins such as lipocalin 2. It is also possible that MSCs secrete other antimicrobial factors in addition to lipocalin 2 and LL-37, and these factors may also participate in the regulation of the antimicrobial effects of MSCs.
In summary, this study demonstrates that MSC-based therapy improves survival, reduces lung injury and enhances bacterial clearance when used as a treatment for bacterial pneumonia in mice. Part of the bacterial clearance effect observed with MSC treatment is mediated by their ability to upregulate lipocalin 2 production in response to inflammatory stimuli such as LPS and TNFα. These findings demonstrate that MSCs can directly enhance the innate immune response to bacterial infection in the lung.
Acknowledgments
The authors would like to thank Xiao Su, MD, PhD, Yuanlin Song, MD and Sandra Brady for their scientific and technical advice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
See Editorial, p 475
NG and AK contributed equally to this work.
Funding This work was supported by the following institutions and grants: NHLBI HL092059 (NG), Parker B Francis Foundation Pulmonary Fellowship (NG) and NHLBI 51856, 51854 (MAM).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.