Article Text

Abstract
Background Tumour necrosis factor (TNF) is upregulated in the alveolar space early in the course of ventilator-induced lung injury (VILI). Studies in genetically modified mice indicate that the two TNF receptors play opposing roles during injurious high-stretch mechanical ventilation, with p55 promoting but p75 preventing pulmonary oedema.
Aim To investigate the effects of selective inhibition of intra-alveolar p55 TNF receptor on pulmonary oedema and inflammation during ventilator-induced lung injury using a newly developed domain antibody.
Methods Anaesthetised mice were ventilated with high tidal volume and given an intratracheal bolus of p55-specific domain antibody or anti-TNF monoclonal antibody (‘pure’ VILI model). As a model of enhanced inflammation, a subclinical dose of lipopolysaccharide (LPS) was included in the intratracheal antibody bolus (LPS+VILI model). Development of lung injury was assessed by respiratory mechanics and blood gases and protein levels in lavage fluid. Flow cytometry was used to determine leucocyte recruitment and alveolar macrophage activation, while lavage fluid cytokines were assessed by ELISA.
Results The ventilation protocol produced deteriorations in respiratory mechanics and gas exchange with increased lavage fluid protein levels in the two models. The p55-specific domain antibody substantially attenuated all of these changes in the ‘pure’ VILI model, while anti-TNF antibody was ineffective. In the LPS+VILI model, p55 blockade prevented deteriorations in respiratory mechanics and oxygenation and significantly decreased neutrophil recruitment, expression of intercellular adhesion molecule 1 on alveolar macrophages, and interleukin 6 and monocyte chemotactic protein 1 levels in lavage fluid.
Conclusions Selective inhibition of intra-alveolar p55 TNF receptor signalling by domain antibodies may open new therapeutic approaches for ventilated patients with acute lung injury.
- ARDS
- cytokine biology
- innate immunity
- lung physiology
- pulmonary oedema
- respiratory measurement
- airway epithelium
- macrophage biology
- neutrophil biology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- ARDS
- cytokine biology
- innate immunity
- lung physiology
- pulmonary oedema
- respiratory measurement
- airway epithelium
- macrophage biology
- neutrophil biology
Key messages
What is the key question?
Does selective inhibition of tumour necrosis factor (TNF) receptor p55 signalling attenuate ventilator-induced lung injury?
What is the bottom line?
Selective inhibition of the p55 TNF receptor within the alveolar space using a domain antibody attenuated pulmonary oedema and inflammation in mouse models of ventilator-induced lung injury, whereas total TNF blockade using a traditional monoclonal antibody was without effect.
Why read on?
This paper reports for the first time that more specific pharmacological modulation of TNF signalling is potentially effective in the treatment of ventilator-induced lung injury.
Introduction
Mechanical ventilation is essential for the treatment of patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS), although this frequently results in the development of ventilator-induced lung injury (VILI).1 While the initiating trigger is unique, the final pathophysiology of VILI is indistinguishable from that of ALI of other aetiologies—that is, characterised by pulmonary oedema and inflammation.2 3 VILI may be particularly amenable to early pharmacological intervention as its iatrogenic nature means that onset occurs in a predictable manner and within the hospital setting.
The proinflammatory cytokine tumour necrosis factor (TNF) has been consistently implicated in the pathogenesis of ALI/VILI, both clinically and in experimental models.3–6 Clinical trials of anti-TNF therapy in patients with sepsis/ALI7–10 have unfortunately shown little or no benefits, and in some cases may have led to increased morbidity. The reasons behind such a lack of benefit are unclear, but are likely to include issues relating to the design of the agents used and also to the complicated biology of TNF signalling. TNF signals through two cell surface receptors, p55 and p75, and recent evidence suggests that signalling through these individual receptors may have differential—even opposing—roles in a number of pathological conditions.11–13 We previously found that biologically active TNF is upregulated within the alveolar space early during the development of VILI.14 Moreover, using TNF receptor knock-out mice, we found that the two receptors of TNF play opposing roles during VILI: p55 receptor knock-out mice were protected while p75 receptor knock-out animals were more susceptible to pulmonary oedema.15 In light of these data, we propose that ‘selective’ inhibition of ‘p55 TNF receptor’ signalling, specifically within the alveolar space, would be clinically much more effective than total TNF inhibition to ameliorate the deleterious effects of mechanical ventilation.
Conventional antibody technologies are not optimal for this purpose for a variety of reasons, perhaps the most important of which being that monoclonal antibodies to this and, in general, other cell surface receptors have a tendency to induce receptor cross-linking, thereby activating rather than inhibiting signalling.16 17 To investigate the possibility of pharmacological inhibition of p55 signalling, we therefore used a novel IgG fragment known as a domain antibody (GlaxoSmithKline, Stevenage, UK) that was selected to specifically bind and inhibit mouse p55 TNF receptor. Domain antibodies (dAbs) comprise the variable domains of either the heavy or light chain, and are thus the smallest functional antigen-binding portions of the IgG molecule.18 19 Domain antibodies offer multiple advantages over conventional antibody technology: they bind monovalently to their targets, avoiding the potential for receptor cross-linking; they lack the Fc portions and do not induce Fc effector responses such as complement-dependent cytotoxicity or antibody-dependent cytotoxicity; their small size (∼12 kD) enables better tissue penetration, delivery at much higher concentrations per unit mass and potentially fewer long-term adverse effects (eg, immunosuppression) due to more rapid excretion; and they can be manufactured to be suitable for local delivery such as inhalation.
In this study we investigated the effects of an intratracheally delivered p55 TNF receptor-targeting dAb in in vivo mouse models of VILI. We found that selective blockade of intra-alveolar p55 signalling reduced alveolar-epithelial barrier permeability and pulmonary inflammation, which suggests potential for new treatments of VILI and ALI.
Methods
Ventilation protocol
Male C57BL6 mice (Charles River, Margate, UK) aged 8–12 weeks were anaesthetised (intraperitoneal ketamine 80 mg/kg and xylazine 8 mg/kg), tracheostomised and ventilated using a custom-made jet ventilator.14 20 21 Airway flow was monitored by a miniature pneumotach in the ventilator system and tidal volume (VT) was calculated by integrating flow during inspiration. The left carotid artery was cannulated for blood pressure (BP) and arterial blood gas (ABG) measurements. Ventilation was then set to produce VILI (‘pure’ VILI model), standardised by inspiratory plateau pressure (Pplateau) of 14 cm H2O equating to VT of ∼22 ml/kg with positive end-expiratory pressure 3 cm H2O, respiratory rate 90/min and oxygen+4% carbon dioxide (to prevent hypocapnia). Immediately afterwards, mice received an intratracheal bolus of either dummy (non-targeting) dAb or p55-specific blocking dAb (25 μg), or either mouse anti-TNF monoclonal antibody (50 μg; TN3-19.12; BD Pharmingen, Oxford, UK) or its isotype control (hamster IgG, BD Pharmingen) in a total volume of 50 μl. We previously demonstrated the efficacy of the monoclonal anti-TNF used (with the same route and dose) to reduce alveolar neutrophil infiltration associated with VILI.22 In a separate series of experiments, a subclinical dose of lipopolysaccharide (LPS, 20 ng; Escherichia coli O111:B4, Source Bioscience, Nottingham, UK) was co-administered intratracheally with the dAb bolus to enhance the inflammatory component of the model. In this LPS+VILI model, slightly lower Pplateau (13 cm H2O) and VT (∼20 ml/kg) were used as described previously21 because LPS was expected to enhance the degree of VILI.
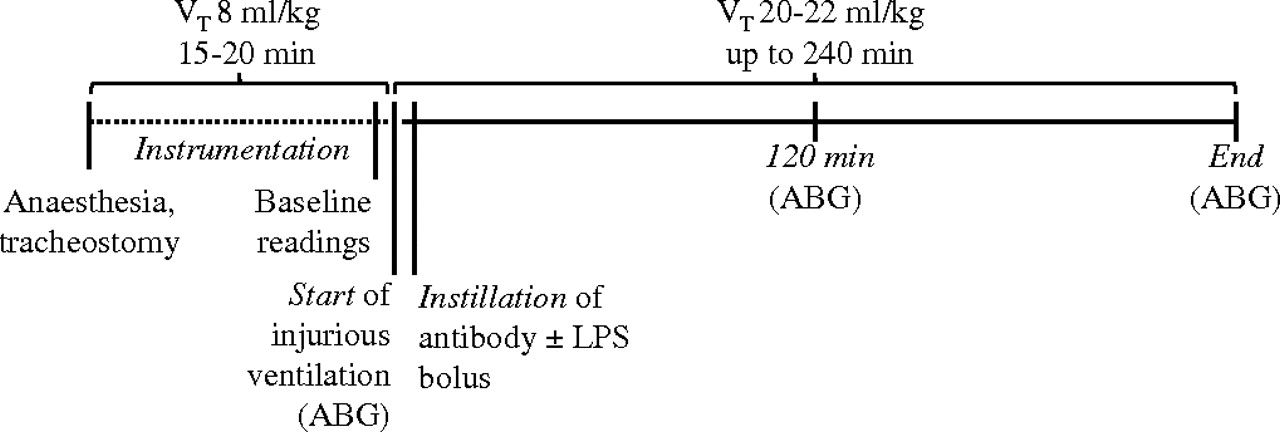
Ventilation using the same constant high VT was continued in all groups for 240 min or until the mean BP dropped below 45 mm Hg.14 Anaesthesia was maintained by bolus administrations of intraperitoneal ketamine:xylazine every 20–25 min. A small number of animals showed very rapid haemodynamic deterioration: in the ‘pure VILI’ model, three mice treated with dummy dAb, one treated with p55-specific dAb, one treated with isotype control and two mice treated with anti-TNF; in the LPS +VILI model, one mouse treated with dummy dAb. These prematurely terminated experiments were excluded from the analyses. Pplateau, respiratory system elastance (Ers) and resistance (Rrs) were determined by the end-inflation occlusion technique20 every 20 min, followed each time by sustained inflation (35 cm H2O, 5 s) to avoid the development of atelectasis. ABG were assessed at predetermined points throughout the protocol (figure 1).
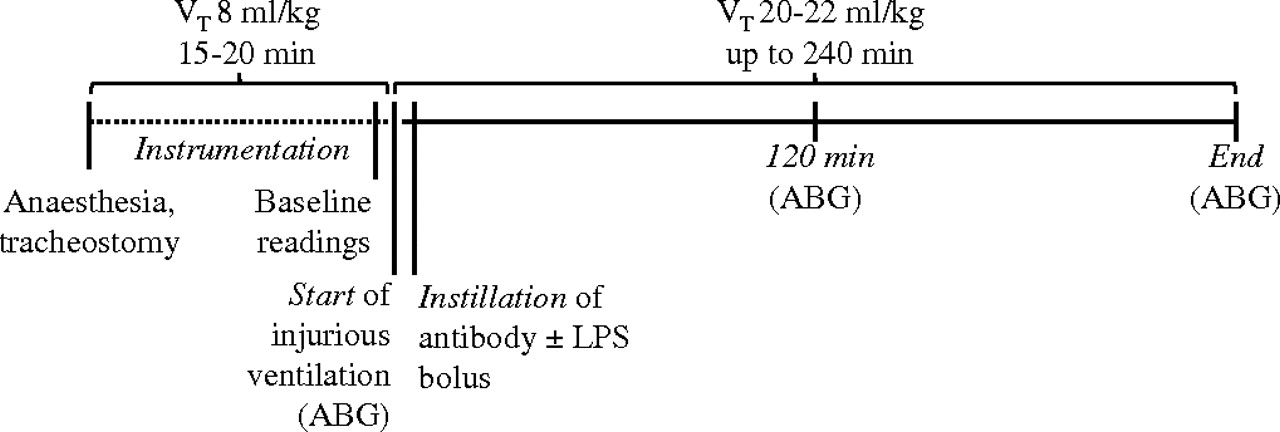
Scheme of ventilation protocol. Mice were anaesthetised, tracheostomised and ventilated with non-injurious ventilation for approximately 15–20 min during instrumentation (dotted line). Injurious ventilation parameters using high tidal volumes (VT) were then introduced, followed immediately by instillation of the antibody of interest. As a model of ventilation-induced lung injury with enhanced inflammation, a subclinical dose of lipopolysaccharide (LPS, 20 ng) was co-administered with the domain antibody bolus. After instillation of the reagents, four sustained inflations (35 cm H2O, 5 s) were applied to help the fluid distribute within the lungs. Respiratory mechanics were assessed every 20 min, followed each time by sustained inflation. Arterial blood gases (ABG) were assessed at predetermined time points (start, 120 min, end).
Lung lavage
Following termination, lung lavage was performed14 and lavage fluid was analysed for protein concentration (Bio-Rad Laboratories, Hemel Hempstead, UK) and, in some cases, for TNF levels by ELISA (R&D Systems, Abingdon, UK). In the LPS+VILI model, levels of interleukin 6 (IL-6), monocyte chemotactic protein 1 (MCP-1), keratinocyte chemoattractant (KC) and macrophage inflammatory protein 2 (MIP-2) in the lavage fluid were evaluated by ELISA (R&D) and cell pellets were used for flow cytometry. Cytokine levels are expressed as total amount (pg) recovered by lavage.
Lung histology
In some experiments, lungs were removed and instilled with 1% low melting point agarose in 4% paraformaldehyde and processed for histological evaluation by H&E staining.
Flow cytometry
In the LPS+VILI model, lung single cell suspensions were prepared from excised lungs by mechanical disruption for flow cytometry analysis, as described previously.23–25 Lung and lavage cells were stained with fluorophore-conjugated anti-mouse antibodies for CD11b, CD11c, Gr-1 (Ly6C/G), F4/80 and intercellular adhesion molecule 1 (ICAM-1) or appropriate isotype-matched controls, and analysed using a FACSCalibur flow cytometer with CellQuest (Becton Dickinson, Oxford, UK) and Flowjo (Tree Star, Ashland, OR, USA) software. Cells were quantified using microsphere counting beads (Caltag Medsystems, Towcester, UK) added to the samples. Activation state of alveolar macrophages was evaluated based on surface ICAM-1 expression.
Statistical analysis
Data are expressed as mean±SD. Statistical comparisons were made by two-way repeated measures ANOVA for time course data or unpaired t tests for end point data using Prism software Version 5.0. A value of p<0.05 was considered significant. For some end point analyses, data were compared with those of non-ventilated mice for qualitative purposes.
Results
Effects of anti-p55 domain antibody on respiratory physiology in the ‘pure’ VILI model
Mechanical ventilation with the high stretch protocol caused a substantial deterioration in lung function in dummy dAb-treated animals, consistent with pulmonary oedema formation. This was represented by a rapidly increasing Pplateau, Ers and Rrs that occurred towards the end of the 4 h ventilation protocol (figure 2A–C). In marked contrast, no deteriorations were seen in respiratory mechanics in the p55 dAb group.

(A) Plateau pressure (Pplateau), (B) respiratory system elastance (Ers), (C) respiratory system resistance (Rrs), (D) mean arterial pressure, (E) arterial oxygen tension (PO2), (F) arterial carbon dioxide tension (PCO2), (G) bicarbonate (HCO3−) and (H) lavage fluid protein were determined in ventilated animals treated intratracheally with either dummy or p55-specific domain antibody (dAb). Changes in Ers and Rrs are expressed as percentage increase in relation to Start (before instillation) values. Pplateau, Ers and Rrs changes over time during ventilation were significantly different between treatment groups (#p<0.05 for interaction between treatment and ventilation time by two-way ANOVA). Arterial PO2 showed a tendency to deteriorate in animals treated with the dummy dAb but not in mice given the p55-specific dAb. Changes in PCO2 over time were significantly different between treatment groups (#p<0.05 for interaction by two-way ANOVA), while changes in bicarbonate levels were similar across groups. Lavage fluid protein was significantly attenuated in the p55-specific dAb group (*p<0.05 by unpaired t tests). The dotted line represents the mean value in non-ventilated control animals (N=8–11 per group).
Arterial oxygen and carbon dioxide tensions (PO2 and PCO2) were similar between the two groups at the start and 120 min after drug instillation (figure 2E,F). By the end of the protocol, oxygenation tended to deteriorate in dummy dAb-treated animals (365±146 mm Hg) while PCO2 increased. In contrast, PO2 was better maintained in the p55 dAb group (440±41 mm Hg) and PCO2 was unchanged from the 120 min level. To assess alveolar epithelial permeability we measured the protein content in lavage fluid. VILI resulted in substantial protein levels in the dummy dAb group compared with non-ventilated controls (2.82±1.73 vs 0.21±0.03 mg/ml), and this was significantly attenuated in mice given the p55 dAb (1.34±0.4 mg/ml, figure 2H). TNF levels in bronchoalveolar lavage fluid evaluated at the end of the ventilation protocol in the dummy dAb group were found to be upregulated (147±144 pg, n=11) compared with untreated mice (less than the assay detection limit of 15 pg/ml).
Lung histology indicated a moderate degree of injury after VILI with dummy dAb treatment, characterised by neutrophil infiltration and proteinaceous material in the alveolar space (figure 3A,B) that was substantially reduced following p55 dAb (figure 3C,D).

Photomicrographs of lung tissue. Sample from dummy domain antibody (dAb)-treated mouse (A, B) showed marked cellularity, alveolar wall thickening (arrow), hyaline membrane formation and intra-alveolar debris (arrowhead) whereas minimal changes were seen in p55 dAb-treated mouse (C, D). H&E stain. Magnification ×100 (A and C), ×400 (B and D).
Effects of anti-TNF monoclonal antibody on respiratory physiology during VILI
Having found that blockade of p55 TNF receptor signalling attenuated VILI, we investigated whether total TNF blockade would be similarly efficacious. To address this, we treated mice with anti-TNF monoclonal antibody (or isotype control) using the same instillation strategy and ventilation protocol as for the dAb experiments.
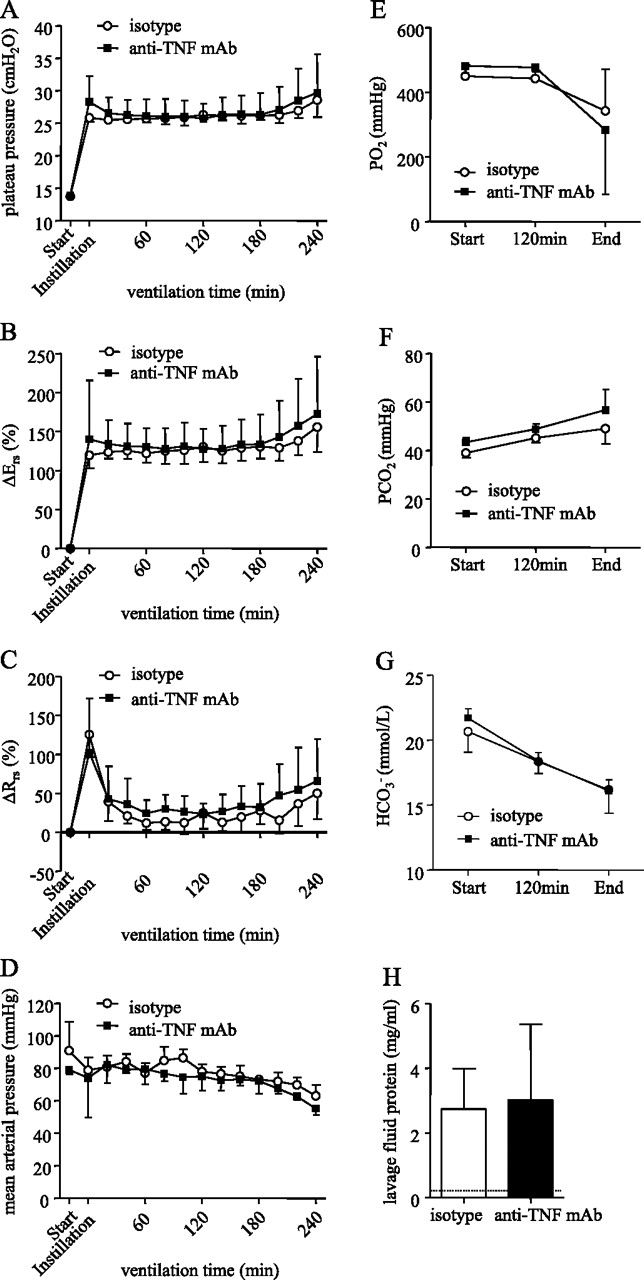
We found that changes in respiratory mechanics in the isotype control-treated group followed a very similar pattern as was observed for the dummy dAb group (figure 4A–C). In marked contrast to the beneficial effect of the p55-specific dAb, however, the anti-TNF monoclonal antibody did not attenuate the changes seen in isotype-treated animals. Similarly, anti-TNF treatment had no beneficial effect on blood gases (figure 4E–G) or alveolar epithelial permeability (figure 4H).

(A) Plateau pressure (Pplateau), (B) respiratory system elastance (Ers), (C) respiratory system resistance (Rrs), (D) mean arterial pressure, (E) arterial oxygen tension (PO2), (F) arterial carbon dioxide tension (PCO2), (G) bicarbonate (HCO3−) and (H) lavage fluid protein were not significantly different between animals treated intratracheally with either isotype or mouse anti-tumour necrosis factor (TNF) monoclonal antibody (mAb). Dotted line represents mean value in non-ventilated control animals (N=6 per group).
Effects of anti-p55 domain antibody on inflammation following LPS+VILI
Finally, we evaluated whether the p55-specific dAb would also have a beneficial effect on pulmonary inflammation during VILI. Our group and others26 have shown that pure mechanical ventilation induces a relatively limited degree of alveolar neutrophil infiltration (within the time frame of ventilation used currently), one of the hallmarks of clinical ALI. We therefore included a ‘subclinical’ dose of LPS (20 ng) mixed with the dummy or p55-specific dAbs at the start of mechanical ventilation to enhance the degree of inflammation within the model. Preliminary studies demonstrated that this dose of LPS did not induce changes in respiratory mechanics with low tidal volume ventilation.
As with the ‘pure’ VILI model, LPS+VILI induced substantial changes in respiratory mechanics which were significantly attenuated by treatment with p55-specific dAb (figure 5A–C). Similarly, administration of the p55 dAb prevented the deterioration of oxygenation (figure 5E) and tended to attenuate the rise in PCO2 (figure 5F). p55 dAb also tended to decrease protein levels in lavage fluid (2.94±1.62 vs 2.06±1.45 mg/ml for dummy and p55 dAb, respectively), although this did not reach statistical significance (figure 5H).

(A) Plateau pressure (Pplateau), (B) respiratory system elastance (Ers) and (C) resistance (Rrs), (D) mean arterial pressure, (E) arterial oxygen tension (PO2), (F) arterial carbon dioxide tension (PCO2), (G) bicarbonate (HCO3−) and (H) lavage fluid protein of dummy domain antibody (dAb)-treated and p55 dAb-treated mice in the lipopolysaccharide + ventilation-induced lung injury (LPS+VILI) model. Changes in Ers and Rrs are expressed as percentage increase in relation to Start values. Changes in Pplateau, Ers, Rrs, arterial PO2 and bicarbonate over time were significantly different between treatment groups (#p<0.05 for interaction between treatment and ventilation time by two-way ANOVA). The p55-specific dAb tended to attenuate the rise in PCO2, but this did not reach statistical significance (p=0.055 for interaction by two-way ANOVA). Lavage fluid protein levels tended to be reduced in p55-specific dAb-treated animals, but not significantly. The dotted line represents the mean value in non-ventilated controls (N=8–11 per group).
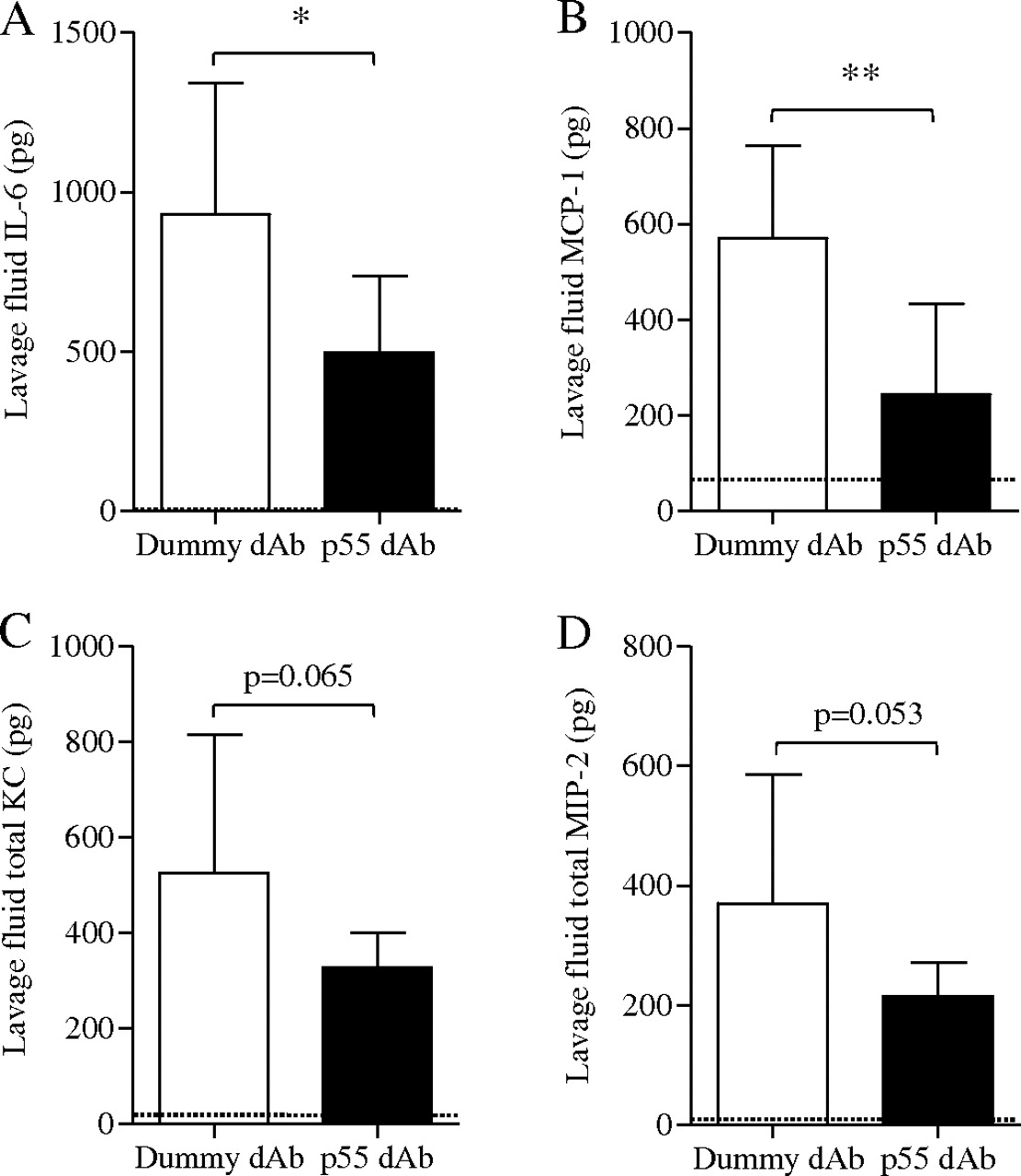
Leucocyte subsets in lung single cell suspension and lavage fluid were identified based on characteristic antigen expression using flow cytometry (figure 6A–C) and their number or activation state was quantitatively assessed. LPS+VILI increased the number of lung-recruited neutrophils compared with non-ventilated controls (2.32±0.57×106 vs 0.13±0.05×106), which was significantly attenuated by the p55-specific dAb (1.25±0.31×106, p<0.01 vs dummy; figure 6A). A similar trend was found with margination of inflammatory subset Gr-1high monocytes, although this was not significant (figure 6A). Transmigration of neutrophils to the alveolar space was also greatly increased by LPS+VILI and substantially attenuated by p55 dAb treatment (figure 6B). Alveolar macrophages in lavage fluid of dummy dAb-treated mice showed increased expression of ICAM-1 compared with non-ventilated controls, indicative of activation,27 which was also reduced in the p55 dAb group (figure 6C). Finally, treatment with p55 dAb significantly decreased IL-6 and MCP-1 in lavage fluid, with similar trends for KC and MIP-2 (figure 7).
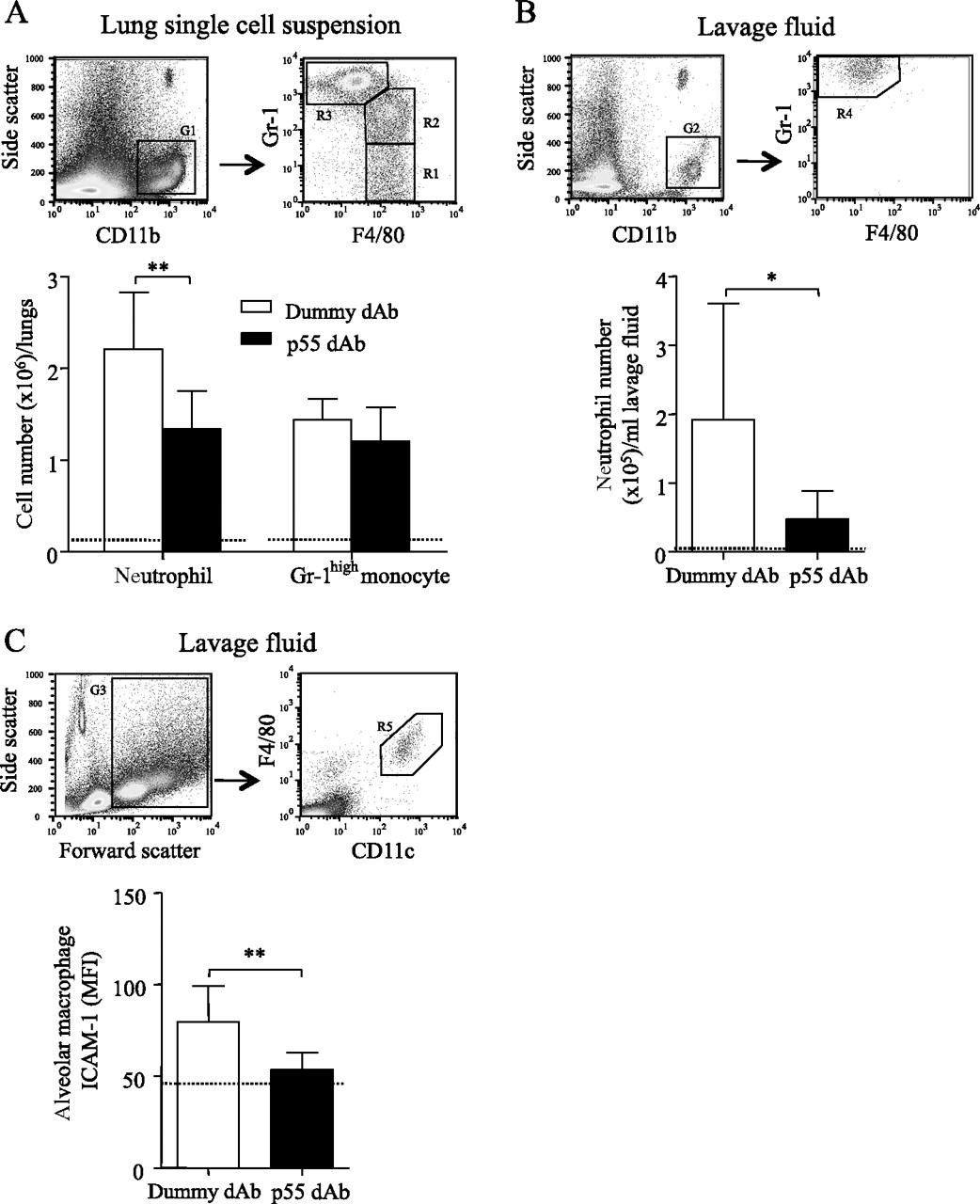
Flow cytometric analysis of leucocyte subsets in the lungs and lavage fluid of dummy domain antibody (dAb)-treated and p55 dAb-treated mice in the lipopolysaccharide + ventilation-induced lung injury (LPS+VILI) model. (A) In lung single-cell suspensions, monocytes were identified as CD11b+, F4/80+ events, and their subsets were defined as either Gr-1low (R1) or Gr-1high (R2), differentiated from CD11b+, F4/80−, Gr-1very high neutrophils (R3). (B) Neutrophils in lavage fluid were similarly identified as CD11b+, F4/80−, Gr-1very high events (R4), whereas monocytes were not present. (C) Alveolar macrophages in lavage fluid were recognised as forward/side scatterhigh, F4/80+, CD11c+ events (R5) and their intercellular adhesion molecule (ICAM-1) expression was quantitatively assessed (mean fluorescence intensity (MFI) after isotype subtraction). The dotted line represents the mean value in non-ventilated control animals. *p<0.05, **p<0.01 by unpaired t test (N=8–11 per group).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of (A) interleukin 6 (IL-6), (B) monocyte chemotactic protein 1 (MCP-1), (C) keratinocyte chemoattractant (KC) and (D) macrophage inflammatory protein 2 (MIP-2) in lavage fluid of dummy domain antibody (dAb)-treated and p55 dAb-treated mice in the lipopolysaccharide + ventilation-induced lung injury (LPS+VILI) model. The dotted line represents the mean value in non-ventilated control animals (undetectable levels for IL-6). *p<0.05, **p<0.01 by unpaired t test (N=8–11 per group).
Discussion
ALI and ARDS are characterised by pulmonary oedema, hypoxaemia and inflammation. While mechanical ventilation is an essential aspect of treatment, this can exacerbate underlying lung injury and cause VILI. It has been suggested that this can progress to systemic inflammation and multiple system organ failure, which is a major cause of death among patients with ARDS.28 29 Although mortality is improved by the use of low tidal volume ventilation in conjunction with other interventions, it is not possible to eliminate completely the consequences of VILI in this manner. An ideal therapeutic agent would therefore attenuate both the respiratory dysfunction which drives the requirement for ventilation and the inflammatory response which may ultimately lead to death. In this study we investigated the use of a novel therapeutic technology—domain antibodies—for the first time in the area of ALI, and showed that intratracheal administration of dAb targeting the p55 TNF receptor attenuated the development of VILI.
Using a mouse model of ‘pure’ VILI we found that deteriorations in respiratory function and alveolar-epithelial barrier permeability were substantially improved when p55-specific blocking dAb was given intratracheally at the start of the ventilation protocol. In complete contrast, intratracheal administration of a monoclonal anti-TNF antibody did not attenuate stretch-induced respiratory dysfunction. In addition, in an LPS+VILI model incorporating a greater degree of pulmonary inflammation, administration of the p55-specific dAb led to decreased levels of the proinflammatory cytokine IL-6 and the monocyte/neutrophil chemoattractant MCP-1 in lavage fluid, and showed strong trends towards decreases in the chemokines MIP-2 and KC. While the precise sources of these inflammatory mediators in VILI remain uncertain, alveolar macrophages are likely contributors and, indeed, we observed that alveolar macrophage activation (surface ICAM-1 expression27) was significantly attenuated by dAb treatment. Presumably as a consequence of this reduced intra-alveolar inflammation, margination and migration of neutrophils within the lung was also attenuated. These data demonstrate clearly that specific blockade of p55 TNF-receptor signalling within the alveolar space attenuates multiple markers of VILI development.
The ventilation parameters used within this study (VT 20–22 ml/kg), although higher than used clinically in humans, would have induced only a moderately high degree of lung stretch in untreated healthy mice as mouse lungs are much more compliant than those of humans.30 However, due to the impact of the intratracheal fluid instillation, such settings were sufficient to induce injury within 4 h of ventilation. While the timeframe of the current experiment is much shorter than patients with ALI would be ventilated, it is comparable with the majority of published rodent studies of VILI and ALI. Sustained inflations and post end-expiratory pressure were used throughout the ventilation protocols as mouse lungs have a very high tendency for atelectasis even at such VT, which could have substantial confounding effects. Since atelectasis was minimised by these recruitment measures, the major component of the observed physiological changes can be ascribed to the development of stretch-induced pulmonary oedema. The dAb was given immediately after setting ventilation parameters in order to allow robust standardisation of the VILI protocol, avoiding the unpredictable influence of instillation on respiratory mechanics (upon which the degree of VILI was standardised). The finding that pulmonary oedema and inflammation during VILI were attenuated under these conditions would indicate that a substantial prophylactic period was unnecessary for dAb efficacy.
From the current data it is not possible to determine precisely why the p55-specific dAb was clearly more effective in attenuating VILI than the anti-TNF monoclonal antibody. One possibility is that greater efficacy of the dAb was related to its biophysical properties—its small size (∼12 kDa compared to ∼150 kDa for IgG) and monovalent target antigen binding allows more potency per unit mass, greater tissue penetration and better biodistribution, and reduced risk of receptor cross-linking and activation compared with conventional antibody technology. Thus, despite the fact that we used a 2× larger amount (in terms of weight) of the monoclonal antibody, the dAb could be delivered at an approximately 6× greater dose than the monoclonal antibody which may simply induce more effective inhibition of TNF receptor binding. This lack of efficacy of anti-TNF treatment to attenuate the physiological manifestations of VILI are somewhat at odds with a previous study showing a positive effect of anti-TNF in a rabbit model of saline lavage followed by injurious ventilation,4 but do reflect the disappointing results from clinical trials in patients with sepsis/ARDS.7–10 31
In addition to the biophysical properties of the respective antibodies, we believe target antigen specificity is vitally important. Despite the large number of studies investigating TNF as a potential therapeutic agent, the complexity of TNF biology remains rather poorly appreciated. It has been assumed that the majority of TNF signalling occurs through the p55 receptor, with p75 playing only an auxiliary role, but it is becoming clearer that the two receptors can signal independently and, in some cases, may exert directly opposite effects to each other.11 15 For example, we previously showed using knock-out mice that signalling through p55 promoted the development of high stretch-induced pulmonary oedema, while signalling through p75 protected against oedema formation during VILI, and that these opposing effects seemed to be independent of TNF-mediated leucocyte recruitment.15 Such was the nature of this opposing signalling that mice devoid of all TNF receptors showed no protection from oedema (a finding consistent with others32). These data from genetically modified animals suggest that non-selective anti-TNF treatment would not be effective however high a dose of antibody we may be able to deliver. Interestingly, in an earlier study we showed that the monoclonal anti-TNF antibody used currently attenuated neutrophilic alveolitis in VILI.22 Similarly, Wolthuis et al found that total blockade of TNF signalling by etanercept, a p75 receptor-Fc fusion protein, failed to attenuate high stretch-induced pulmonary oedema but had some positive effects on inflammation.33 Taken together, these data suggest that, while treatment designed to block TNF per se may be effective in reducing pulmonary inflammation associated with VILI, it has little impact on pulmonary oedema, presumably as any beneficial effects of signalling through the p75 receptor on pulmonary fluid balance are inhibited. The nature of these beneficial effects of p75 signalling remains unclear, but we speculate that it may involve actin cytoskeleton rearrangements34 35 or prevention of epithelial/endothelial apoptosis.12
In conclusion, we have shown for the first time that specific and selective pharmacological inhibition of intra-alveolar TNF receptor p55 signalling by a dAb can ameliorate pulmonary oedema and inflammation during VILI in mice. Targeting intra-alveolar TNF receptor p55 signalling may open new therapeutic approaches for ventilated patients with ALI.
Acknowledgments
The authors thank A C Waite for her assistance with the flow cytometry measurements.
References
Footnotes
Funding This study was supported by grants from GlaxoSmithKline (GSK) (COL028224) and the Wellcome Trust (#081208). GSK have a financial interest in the use of domain antibodies, including those targeting p55 TNF receptor, in the treatment of pulmonary and other diseases. GSK were involved in the initial design of the study and contributed to the study report. However, GSK had no involvement in collection, analysis and interpretation of data or in the decision to submit the report for publication. GSK provided salary funds for one of the authors (SB) and three others (PJM, RdeW and AB) are employees of GSK. GSK also provided funds for animal and consumable costs. The Wellcome Trust provided support of infrastructure for the animal experiments.
Competing interests PJM, RdW and AB are employed by and hold stock in GSK. SB, MW and MT have no competing interests.
Ethics approval All protocols were approved by the ethical review board of Imperial College London as well as the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and carried out under the authority of the UK Home Office in accordance with the Animals (Scientific Procedures) Act 1986.
Provenance and peer review Not commissioned; externally peer reviewed.