Article Text

Abstract
Background The development of organ fibrosis after injury requires activation of transforming growth factor β1 which regulates the transcription of profibrotic genes. The systemic administration of a proteasomal inhibitor has been reported to prevent the development of fibrosis in the liver, kidney and bone marrow. It is hypothesised that proteasomal inhibition would prevent lung and skin fibrosis after injury by inhibiting TGF-β1-mediated transcription.
Methods Bortezomib, a small molecule proteasome inhibitor in widespread clinical use, was administered to mice beginning 7 days after the intratracheal or intradermal administration of bleomycin and lung and skin fibrosis was measured after 21 or 40 days, respectively. To examine the mechanism of this protection, bortezomib was administered to primary normal lung fibroblasts and primary lung and skin fibroblasts obtained from patients with idiopathic pulmonary fibrosis and scleroderma, respectively.
Results Bortezomib promoted normal repair and prevented lung and skin fibrosis when administered beginning 7 days after the initiation of bleomycin. In primary human lung fibroblasts from normal individuals and patients with idiopathic pulmonary fibrosis and in skin fibroblasts from a patient with scleroderma, bortezomib inhibited TGF-β1-mediated target gene expression by inhibiting transcription induced by activated Smads. An increase in the abundance and activity of the nuclear hormone receptor PPARγ, a repressor of Smad-mediated transcription, contributed to this response.
Conclusions Proteasomal inhibition prevents lung and skin fibrosis after injury in part by increasing the abundance and activity of PPARγ. Proteasomal inhibition may offer a novel therapeutic alternative in patients with dysregulated tissue repair and fibrosis.
- Bortezomib/Fibrosis/PPARγ/Proteasomal/TGF-β1
- ARDS
- cytokine biology
- sleep apnoea
- airway epithelium
- oxidative stress
- pulmonary oedema
Statistics from Altmetric.com
- Bortezomib/Fibrosis/PPARγ/Proteasomal/TGF-β1
- ARDS
- cytokine biology
- sleep apnoea
- airway epithelium
- oxidative stress
- pulmonary oedema
Key messages
What is the key question?
Can the systemic administration of the proteasomal inhibitor bortezomib prevent the development of lung fibrosis after injury is established?
What is the bottom line?
Bortezomib protects against the development of fibrosis in the lung and the skin by inhibiting transforming growth factor β1-mediated transcription.
Why read on?
Bortezomib, a medication in widespread clinical use, may offer a therapeutic alternative for patients with lung fibrosis.
Introduction
After injury, the recovery of normal tissue function can be prevented or delayed by the development of fibrosis.1 In animal models, activation of the cytokine transforming growth factor β1 (TGF-β1) is required and sufficient for the development of fibrosis in the lung and other organs.2 In the lung, the intratracheal administration of bleomycin results in acute lung injury that peaks 3–5 days after the administration of bleomycin and is followed by TGF-β1-dependent lung fibrosis.2 Active TGF-β1 binds to specific membrane receptors inducing signalling cascades that transcriptionally regulate myofibrobast differentiation, collagen expression and endothelial/epithelial to mesenchymal cell transition.3 The transcriptional programme activated by TGF-β1 is therefore an attractive therapeutic target for the prevention of organ fibrosis after injury.2
In addition to its role in protein turnover, the ubiquitin/proteasome system plays a critical role in the modulation of critical cellular signalling pathways.4 Bortezomib is a proteasomal inhibitor approved for clinical use in the treatment of multiple myeloma and mantle cell lymphoma.5 6 In animal models, bortezomib and other proteasome inhibitors can prevent fibrosis in the liver, kidney, bone marrow and heart, suggesting that it acts to inhibit a common pathway involved in organ fibrosis.7–10 In this study we sought to determine whether the administration of bortezomib could prevent lung and skin fibrosis induced by bleomycin by inhibiting TGF-β1-mediated transcription.
Methods
Animals and administration of bleomycin and bortezomib
The protocols for the use of animals were approved by the Northwestern University Animal Care and Use Committee. The protocol for bleomycin lung injury/fibrosis has been described elsewhere (see online supplement for details).11 After conducting pilot experiments using different dosing schedules of bortezomib (details in the online supplement), wild-type C57BL/6 mice were treated with intratracheal bleomycin (0.075 IU/mouse) followed 7 and 14 days later by bortezomib (120 μg/kg intraperitoneally) or saline and lung fibrosis was measured on day 21. For the skin fibrosis experiments, 6–8-week-old female BALB/c mice were treated with filter-sterilised bleomycin (20 μg/mouse, Mayne Pharma, Paramus, New Jersey, USA) or saline subcutaneously daily (27 gauge needle) into a shaved area of skin on the back of the animal. Bortezomib (400 μg/kg intraperitoneally) or vehicle was begun 7 days after the first dose of bleomycin and administered twice weekly until the animals were killed on day 40.
Cells and reagents
Normal human lung fibroblasts were obtained from Lonza (Basel, Switzerland). The cells were grown to 70% confluence for all conditions and were discarded after passage 5. Antibodies used include αSMA (R&D Systems, Minneapolis, Minnesota, USA), fluorescent anti-mouse antibody (Invitrogen, Carlsbad, California, USA), p-Smad3 (Cell Signaling, Boston, Massachusetts, USA), Smad1,2 3 (total Smad), CTGF and PPARγ (Santa Cruz, Santa Cruz, California, USA), collagen I (SouthernBiotech, Birmingham, Alabama, USA) and actin and tubulin (Sigma-Aldrich, St Louis, Missouri, USA). SBE-luciferase and PPRE-luciferase reporters have been previously described.12 13 Measurement of luciferase activity (Promega Dual-Luciferase Reporter Assay System) was performed as previously described (see details in online supplement).14
Real-time quantitative PCR (RT-qPCR)
Real-time quantitative PCR was performed as previously described according to published guidelines and specific mRNA expression was normalised to that of the mitochondrial gene RPL19.15 Detailed protocols and primer sequences are given in the online supplement.
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence were performed as previously described (see details in the online data supplement).16
Measurement of active TGF-β1
Active TGF-β1 was measured from bronchoalveolar lavage (BAL) fluid in duplicate using the TGFβ1 Emax ImmunoAssay System (ELISA) according to the manufacturer's protocol (Promega, Madison, Wisconsin, USA). This assay only measures TGF-β1 that has been cleaved and is biologically active.17
Histology and measurement of lung collagen
Lung and skin histology were performed as previously described.11 18 Lung collagen was measured using a modification of a previously described method.19 Details are provided in the online supplement.
Lentiviral PPARγ shRNA
The pLKO.1 vector was used to express shRNA targeting PPARγ1 as described previously.19 The following sequence was used: 5′CCGGCTGGCCTCCTTGATGAATAAACTCGAGTTTATTCATCAAGGAGGCCAGTTTTT'3. The control shRNA was supplied by Sigma. Stable cell lines were generated by Virapower (Invitrogen) lentiviral infection using the 293FT packaging cell line and puromycin selection. Forty-eight hours after 293FT transfection, medium containing virus was supplemented with 8 μg/ml polybrene (Sigma-Aldrich) for cell line infection and applied to normal human lung fibroblasts.
Clinical specimen collection
The collection of clinical data and specimens was approved by the Northwestern University Institutional Review Board. BAL fluid from 10 patients with a clinical and radiological diagnosis of lung fibrosis and eight control patients was used.14 The clinical details of the patients are given in the online supplement. Normal human lung fibroblasts were transiently transfected with the SBE-luciferase plasmid and incubated for 24 h with BAL fluid obtained from 10 patients with lung fibrosis or control patients without evidence of parenchymal lung disease in the presence or absence of bortezomib (200 nM).14 Samples of lung fibroblasts from fibrotic lungs and control lungs were cultured and stored as previously described.20 Samples of skin fibroblasts were obtained by skin biopsy of an affected and unaffected area of the forearm of a patient with scleroderma as previously described.21
Statistical analysis
The data were analysed in Prism 4 (GraphPad Software Inc, La Jolla, California, USA). All data are shown as means with standard errors of the mean. Statistical significance was determined by ANOVA; when the ANOVA revealed a significant difference, individual differences were explored using t tests with the Bonferroni or Dunnett correction for multiple comparisons. p Values <0.05 were considered statistically significant.
Results
Bortezomib inhibits bleomycin-induced fibrosis in lung and skin
Similar to previous reports, we observed that >70% of mice died when high- or low-dose bortezomib (400 or 120 μg/kg intraperitoneally, respectively) was administered before or concomitant with bleomycin.22 We did not observe excess mortality in mice when bortezomib was given 7 days or more after bleomycin administration (a description of the dosing schedules employed is given in the online supplement). The lowest dose of bortezomib used (120 μg/kg intraperitoneally) was sufficient to acutely inhibit the chymotrypsin-like activity of the proteasome in red blood cells and serum of control mice (see figure S1 in online supplement). Mice treated with bortezomib (120 μg/kg intraperitoneally) 7 and 14 days after bleomycin administration were harvested for assessment of lung collagen assessed by histological examination of trichrome-stained lung sections, picrosirius red collagen precipitation (figures 1A and C) and immunoblotting using an antibody against type I collagen (see figure S2f in online supplement). Fibrosis was also attenuated in mice treated with the single dose of bortezomib on day 7 (see figure S3 in online supplement) and in a limited number of mice treated with bortezomib (120 μg/kg intraperitoneally) 14 and 21 days after bleomycin administration and harvested for assessment of fibrosis at day 28 (see figure S4 in online supplement).

Bortezomib abrogates pulmonary and skin fibrosis induced by bleomycin in mice. (A) Mice (C57BL/6, male) were treated with intratracheal bleomycin or saline (0.075 IU). Seven and 14 days later the mice were treated with bortezomib (120 μg/kg intraperitoneally) or saline. The mice were killed on day 21 and the lungs were examined by trichrome staining. (B) A separate group of mice (BALB/c) were treated with daily subcutaneous injections of bleomycin (20 μg) in the same region of the skin on their back and bortezomib (400 μg/kg intraperitoneally twice weekly) or saline was begun 7 days later. After 4 weeks, H&E-stained skin sections of the treated skin were obtained. The thickest regions of skin for each treatment are shown. (C) Total lung collagen was quantified by picrosirius red collagen precipitation and (D) skin thickness was measured microscopically (n≥8 (lung) and n=5 (skin) for each treatment arm). p Values for comparisons between groups are indicated in italics above the bars.
We also examined the effect of bortezomib in a murine model of scleroderma skin fibrosis.23 Compared with saline, bortezomib (400 μg/kg intraperitoneally twice weekly) substantially attenuated skin fibrosis in bleomycin-treated mice as assessed by histology and quantification of dermal thickness (figures 1B and 1D).
Bortezomib inhibits TGF-β1-induced gene expression in fibroblasts by preventing Smad-mediated transcription
We transfected primary normal human lung fibroblasts with a Smad-responsive luciferase reporter construct.12 Bortezomib resulted in a dose-dependent inhibition of Smad-mediated transcription in response to recombinant TGF-β1 (figure 2A). At a dose of 200 nM, bortezomib was as effective as the ALK5 receptor inhibitor SB431542 (10 μM) (figure 2A). No significant cell death was observed at the doses of bortezomib used (see figure S5 in online supplement). Following TGF-β1 stimulation, bortezomib caused a non-significant increase in basal and phosphorylated Smad3 levels in whole cell lysates (figure 2B) and did not affect nuclear levels of phosphorylated Smad3 (figure 2C).

Bortezomib does not inhibit transforming growth factor β1 (TGF-β1)-induced Smad3 phosphorylation or nuclear translocation but inhibits Smad-mediated transcription. (A) Primary cultures of normal human lung fibroblasts were transfected with a plasmid containing SBE-luciferase and 24 h later treated with TGF-β1 (5 ng/ml) with or without bortezomib. SBE-luciferase activity was measured 24 h later. Treatment with the ALK-5 inhibitor SB431542 (10 μM) is shown as a control. p<0.0001 for difference in dose determined by one-way ANOVA; Bonferroni-corrected p<0.05 for comparisons between TGF-β1 alone and all other conditions. (B, C) Primary cultures of normal human lung fibroblasts were grown to 70% confluence and incubated with TGF-β1 (5 ng/ml) with or without bortezomib (0.2 μM). The level of phosphorylated Smad3 was measured in (B) total cell lysates and (C) nuclear extracts using total Smad3 and RNA polymerase II (RNA Pol II) as loading controls, respectively. Phosphorylated-Smad3 expression was quantified using densitometry. p Values for comparisons are indicated in italics above the bars (N ≥3 for all measures).
In normal human lung fibroblasts treated with recombinant TGF-β1, the increase in mRNA and protein levels of the TGF-β1 target genes α-smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF) were attenuated by treatment with bortezomib (200 nM) (figures 3A and 3B). The suppression of TGF-β1-mediated transcription of mRNA encoding CTGF and another TGF-β1 target gene plasminogen activator-1 (PAI-1) persisted for at least 6 days after the removal of bortezomib from the culture medium (figure 3C). This was associated with a persistent reduction in the proteasomal activity of the cell lysates (see figure S3 in online supplement).
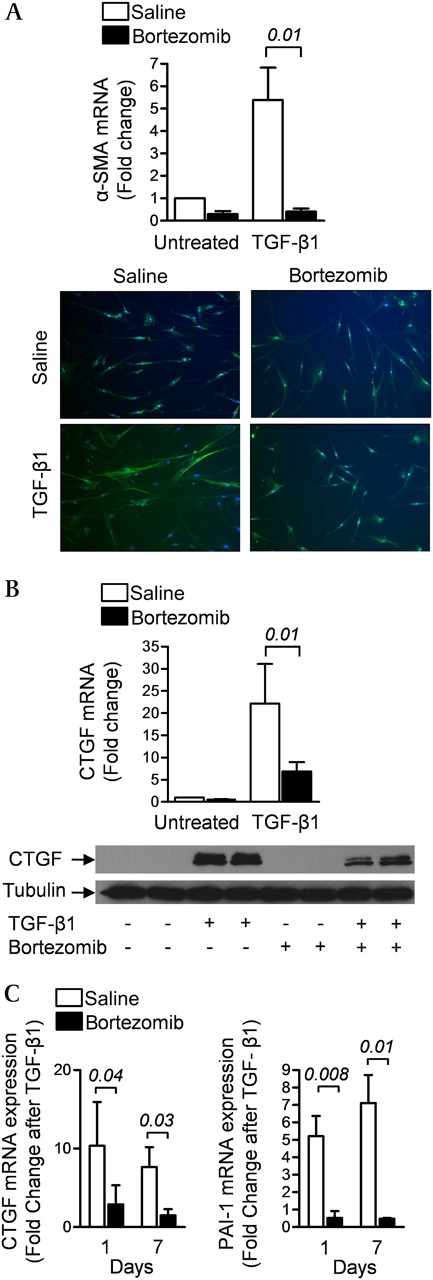
Bortezomib inhibits transforming growth factor β1 (TGF-β1)-induced gene expression in primary normal human lung fibroblasts. (A) Early passage primary cultures of normal human lung fibroblasts were grown to 70% confluence, serum-starved for 24 h and incubated with TGF-β1 (5 ng/ml) with or without bortezomib (200 nM). After 24 h, α-smooth muscle actin (α-SMA) mRNA and protein were measured using RT-qPCR and immunohistochemistry, respectively. (B) Identically treated cells were harvested for measurement of connective tissue growth factor (CTGF) mRNA and protein expression using RT-qPCR and immunoblotting, respectively. (C) Normal human lung fibroblasts were treated with bortezomib (200 nM) for 24 h, cultured in media without bortezomib for 5 days, treated with TGF-β1 (5 ng/ml) in the absence of serum and 24 h later RNA was harvested for measurement of mRNA encoding the TGF-β1 target genes CTGF and plasminogen activator-1 (PAI-1). The number of days after the single dose of bortezomib is indicated (N ≥3 for all conditions). p Values are indicated in italics above the bars.
Bortezomib increases PPARγ abundance and activity in lung fibroblasts
The apparent half-life of PPARγ, a repressor of Smad-mediated transcription, in normal human lung fibroblasts was measured in the presence or absence of bortezomib.24 Bortezomib increased the protein abundance and half-life of PPARγ in the presence and absence of TGF-β1 (figures 4A and 4B). Treatment with bortezomib prevented the TGF-β1-mediated repression of PPARγ transcription as measured using a PPRE-luciferase reporter assay (figure 4C) and the transcription of the PPARγ target gene fatty acid binding protein 4 (FABP4) (figure 4D) (positive (rosiglitazone) and negative (GW9662) controls in figure S6 in online supplement). Wild-type animals were also treated with bortezomib (120 μg/kg intraperitoneally) and 24 h later lung homogenates were immunoblotted using an antibody against PPARγ. Treatment with bortezomib resulted in an increase in total lung levels of PPARγ (figure 4E).

Bortezomib increases PPARγ abundance and activity in normal human lung fibroblasts. (A) Primary normal human lung fibroblasts were cultured in the presence of cycloheximide (10 μg/ml) with and without bortezomib (200 nM) and the protein abundance of PPARγ was assessed by immunoblotting at the indicated times. (B) The same experiment to that described in (A) was performed in cells treated with transforming growth factor β1 (TGF-β1, 5 ng/ml). (C) Primary normal human lung fibroblasts were infected with a plasmid containing PPARγ response element (PPRE)-luciferase. Twenty-four hours after transfection the cells were treated with TGF-β1 (5 ng/ml) with or without bortezomib and PPRE-luciferase activity was measured 24 h later. (D) Primary normal human lung fibroblasts were treated with TGF-β1 (5 ng/ml) with or without bortezomib and the mRNA level of the PPARγ target gene FABP4 was measured 24 h later by RT-qPCR. (E) Bortezomib (120 μg/kg intraperitoneally) was administered to mice and 24 h later lung homogenates were immunoblotted using an antibody against PPARγ. p Values are indicated in italics above the bars (N ≥3 for all measures).
We reasoned that, if the inhibition of TGF-β1-mediated transcription by bortezomib is in part due to an increase in PPARγ, then PPARγ agonists should potentiate and the loss of PPARγ should attenuate the effects of bortezomib. We treated normal human lung fibroblasts with a dose of bortezomib that minimally inhibited TGF-β1-mediated transcription (10 nM, figure 5A), rosiglitazone (10 μM) or the two together. Compared with either agent alone, the combination of low-dose bortezomib with rosiglitazone significantly attenuated the TGF-β1-mediated increase in CTGF mRNA (figure 5A). We used lentiviral shRNA to generate a line of normal human lung fibroblasts with a stable knockdown of PPARγ (or a control lentivirus) and treated these cells with TGF-β1 in the presence or absence of bortezomib. The inhibition of TGF-β1-mediated transcription of CTGF by bortezomib was significantly attenuated in the PPARγ knockdown cells (figure 5B).

Bortezomib modulates fibrotic gene expression via PPARγ. (A) Primary normal human lung fibroblasts were incubated with transforming growth factor β1 (TGF-β1, 5 ng/ml) with or without low-dose bortezomib (10 nM) or rosiglitazone (10 μM) and 24 h later connective tissue growth factor (CTGF) mRNA expression was measured using RT-qPCR. (B) Primary normal human lung fibroblasts were stably transfected with a shRNA against PPARγ or a control lentivurus and PPARγ levels were measured by immunoblotting (densitometry shown to the right of the blot). These cells were treated with TGF-β1 (5 ng/ml) with or without bortezomib (200 nM) and CTGF mRNA was measured 24 h later (RT-qPCR). p Values are indicated in italics above the bars. In addition, p=0.026 for comparison between control transfected cells treated with TGF-β1 or TGF-β1 and bortezomib and p=0.12 for comparison between PPARγ and control shRNA cells treated with TGF-β1 alone.
Bortezomib inhibits the autocrine release of TGF-β1
Normal human lung fibrblasts were treated with TGF-β1 in the presence or absence of bortezomib and TGF-β1 mRNA was measured. The autocrine induction of TGF-β1 was inhibited by bortezomib (figure 6A). To determine whether this mechanism might be important in vivo, BAL fluid levels of active TGF-β1 were measured 14 days after the administration of bleomycin in mice that were treated on day 7 with either bortezomib (120 μg/kg) or saline. The increase in active TGF-β1 in BAL fluid observed 14 days after bleomycin was attenuated in mice that received a single dose of bortezomib at day 7 (figure 6B).

Bortezomib inhibits transforming growth factor β1 (TGF-β1) autocrine expression. (A) Primary normal human lung fibroblasts were incubated with TGF-β1 (5 ng/ml) with or without bortezomib (200 nM) and 24 h later TGF-β1 mRNA expression was measured using RT-qPCR. (B) Intratracheal bleomycin (0.075 IU) or saline was instilled into mice and intraperitoneal bortezomib (120 μg/kg) or saline was administered 7 days later. Bronchoalveolar lavage was performed 14 days after bleomycin treatment for measurement of the level of active TGF-β1 by ELISA. p Values are indicated in italics above the bars.
Bortezomib inhibits profibrotic gene expression in biological samples from patients with pulmonary fibrosis and scleroderma
We examined the effect of bortezomib on the profibrotic gene transcription induced by BAL fluid samples from patients with lung fibrosis. Consistent with our previous report, higher levels of active TGF-β1 were seen in the BAL fluid from patients with lung fibrosis than controls (see figure S7 in online supplement).14 The administration of BAL fluid from 9 out of 10 patients with lung fibrosis increased SBE-luciferase activity in normal human lung fibroblasts and, for each of these nine patients, the increase was inhibited by bortezomib (figure 7A). We then compared the effect of bortezomib on TGF-β1-induced gene transcription in skin fibroblasts isolated from regions of fibrotic or normal skin from a patient with scleroderma.21 Baseline CTGF and PAI-1 mRNA levels were higher in fibroblasts isolated from regions of fibrotic skin than in those from normal skin. In both normal and fibrotic skin fibroblasts the TGF-β1-mediated transcription of these genes was significantly inhibited by bortezomib (figure 7B). Lastly, we examined the effect of bortezomib on TGF-β1-induced gene transcription in primary lung fibroblasts isolated from three patients with lung fibrosis. Both basal and TGF-β1-induced transcription of CTGF and PAI-1 were inhibited by bortezomib in these cells (figure 7C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bortezomib inhibits profibrotic gene expression in biological samples obtained from patients with lung fibrosis or scleroderma. (A) Primary cultures of normal human lung fibroblasts transfected 24 h earlier with a plasmid containing SBE-luciferase were incubated with bronchoalveolar lavage fluid obtained from 10 patients with lung fibrosis or eight patients without evidence of lung fibrosis with or without bortezomib (200 nM). SBE-luciferase activity was measured 24 h later. (B) Primary cultures of skin fibroblasts obtained from a region of normal or fibrotic skin from a patient with scleroderma and (C) primary lung fibroblasts cultured from three patients (Pts 1, 2 and 3) with lung fibrosis were treated with transforming growth factor β1 (TGF-β1, 5 ng/ml) with or without bortezomib or bortezomib alone and the levels of connective tissue growth factor (CTGF) and plasminogen activator-1 (PAI-1) mRNA were measured using RT-qPCR. p Values are indicated in italics above the bars.
Discussion
This study shows that the systemic administration of the proteasomal inhibitor bortezomib to mice beginning 7 days after bleomycin administration prevented lung and skin fibrosis. In primary normal human lung fibroblasts, bortezomib inhibited the TGF-β1-mediated transcription of profibrotic genes downstream of the phosphorylation and nuclear translocation of Smads. Furthermore, bortezomib inhibited Smad-mediated transcription induced by BAL fluid from patients with lung fibrosis and prevented TGF-β1-induced gene transcription in skin and lung fibroblasts from patients with scleroderma and lung fibrosis, respectively. A bortezomib-induced increase in the abundance and activity of PPARγ is an important mechanism contributing to this response.
In animal models, proteasomal inhibitors have been reported to ameliorate liver steatosis/fibrosis,7 myelofibrosis,8 cardiac fibrosis9 and renal fibrosis.10 In these reports, several distinct mechanisms were postulated to explain the antifibrotic effects of proteasomal inhibition including the induction of hepatic stellate cell apoptosis, activation of NF-κB signalling and activation of matrix metalloproteinases. None of these mechanisms accounts for the broad antifibrotic effects of proteasomal inhibition in response to divergent profibrotic stimuli in multiple organs. Our finding that bortezomib inhibits TGF-β1-mediated transcription provides a common mechanism that is consistent with all of these findings.
Consistent with previous reports, we observed that bortezomib did not prevent the phosphorylation and nuclear translocation of Smad3 in response to TGF-β1 in normal human lung fibroblasts.22 Despite this, the activity of a Smad reporter and the expression of TGF-β1 target genes were inhibited in cells treated with bortezomib. These findings suggest that bortezomib exerts its antifibrotic effects at the level of Smad-mediated transcription. The transcriptional response to activated Smads is controlled by both transcriptional co-activators (eg, p300) and co-repressors. The latter include PPARγ, SnoN and Ski.3 25 PPARγ is a nuclear hormone receptor and transcription factor essential for normal adipogenesis and glucose homeostasis that can be activated by endogenous lipids and eicosanoids or thiazolidinedione antidiabetic drugs such as rosiglitazone.26 We have previously reported that PPARγ inhibits TGF-β1 signalling and target gene expression in fibroblasts by acting as a co-repressor of Smad-mediated transcription.25 In this study we found that bortezomib increased the half-life and transcriptional activity of PPARγ. In fibroblasts harbouring a stable knockdown of PPARγ, the effect of bortezomib on TGF-β1-mediated transcription was significantly diminished. Furthermore, bortezomib acted synergistically with the PPARγ agonist rosiglitazone to inhibit TGF-β1-mediated transcription. Collectively these data suggest that, along with other mechanisms, an increase in the abundance and activity of PPARγ contributes to the inhibition of TGF-β1-mediated transcription by bortezomib.
Similar to reports describing the pharmacodynamics of bortezomib in humans, we observed only modest reductions in serum and red blood cell proteasomal activity 3 h after administering the drug.27 Nevertheless, we observed a profound antifibrotic effect when the drug was administered once weekly. This finding is similar to the results obtained in clinical trials using a weekly dosing schedule for the treatment of haematological malignancies.28 It has been speculated that differential clearance of bortezomib from the serum and tissues explains the discrepancy between the kinetics of recovery of proteasomal activity in the blood and the frequency of dosing required to achieve an antineoplastic effect. Our observation that treatment of normal human lung fibroblasts with a single dose of bortezomib inhibited intracellular proteasomal activity and TGF-β1-mediated transcription for up to 7 days supports this hypothesis. Furthermore, TGF-β1 has been reported to induce its own transcription and release from fibroblasts in an autocrine loop.29 We found that bortezomib inhibited the autocrine production of TGF-β1 in vitro and in vivo. We speculate that both the prolonged effect of bortezomib intracellularly and interruption of the autocrine production of TGF-β1 explain its antifibrotic effects at the low doses and less frequent dosing schedules we employed.
The PPARγ agonist rosiglitazone provides modest protection against bleomycin-induced lung fibrosis in mice when administered 3 days before bleomycin.30 This contrasts with the nearly complete protection we observed in mice treated with bortezomib 7 or 14 days after bleomycin. We speculate this difference might result from accelerated proteasomal degradation of PPARγ induced by treatment with rosiglitazone and other thiazolidinediones.31 Consistent with this mechanism, we observed a synergistic inhibition of TGF-β1-mediated transcription in mice treated with a combination of rosiglitazone and bortezomib; the concentration of bortezomib required to completely suppress TGF-β1-mediated transcription was 20-fold lower when it was administered in combination with rosiglitazone.
Fineschi et al reported that bortezomib failed to protect against lung fibrosis in bleomycin-exposed mice; however, these investigators administered bortezomib before or concomitant with the administration of bleomycin.22 Consistent with their findings, we observed excess mortality when bortezomib and bleomycin were administered concomitantly. As the administration of bleomycin results in acute lung injury that peaks 3–5 days later and slowly resolves thereafter, these findings may provide insight into the sporadic reports of patients who have developed pulmonary toxicity, including fatal interstitial pneumonias, while receiving twice weekly bortezomib for haematological malignancies.32–34 While speculative, it may be that the fatal pulmonary events associated with bortezomib resulted from its administration to patients with untreated pneumonia. In support of this hypothesis, therapeutic guidelines developed in response to the reports of bortezomib-related pulmonary complications recommend avoiding bortezomib in patients with evidence of pneumonia. The implementation of these guidelines has been associated with a reduction in bortezomib-related pulmonary toxicity.27 35
Our finding that bortezomib is effective when started 7 days after bleomycin administration is in sharp contrast to other pharmacological therapies that prevent bleomycin-induced lung fibrosis. For example, treatment of mice with imatinib (an inhibitor of Smad-independent signalling by TGF-β1) prior to the administration of bleomycin prevented the subsequent development of lung fibrosis36 but was ineffective when administered after bleomycin.37 A recent clinical trial of imatinib in patients with idiopathic pulmonary fibrosis failed to show improvement in mortality or in lung physiological parameters.38 Based on these and other data, some investigators have advocated that only drugs that are effective in preventing fibrosis when administered after bleomycin should be considered for clinical development.39
Conclusions
Bortezomib, a drug with a well-defined and acceptable toxicity profile and clear mechanism of action,5 6 prevents bleomycin-induced lung fibrosis in mice. It differs from other therapeutic strategies shown to inhibit bleomycin-induced lung fibrosis as it was effective when given after bleomycin-induced acute lung injury had peaked and TGF-β1 was active in the lung. The administration of bortezomib was similarly effective in preventing bleomycin-induced skin fibrosis. Bortezomib effectively inhibited TGF-β1 signalling in primary normal human lung fibroblasts and in fibroblasts from the lungs and skin of patients with lung fibrosis or scleroderma, respectively, and inhibited the profibrotic activity induced by BAL fluid from patients with lung fibrosis. Our results suggest that the bortezomib-mediated increase in the abundance and activity of the TGF-β1 transcriptional repressor PPARγ contributes to its antifibrotic activity. Given the dearth of proven antifibrotic therapies, clinical trials assessing the efficacy of bortezomib alone or in combination with PPARγ agonists in patients with tissue fibrosis may be justified.
Acknowledgments
The authors thank Dr Joan Massagué for the SBE-luciferase construct. The PPRE-luciferase plasmid was generated by Dr Ronald Evans and kindly provided to us by Dr Christopher Glass. The authors thank Dr Aaron Ciechanover for his assistance in the development and review of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
GMM and GRSB contributed equally.
Funding This work was supported by NIH ES015024, ES 013995, HL092963 and HL071643 the Veterans Administration and the Northwestern Memorial Foundation.
Competing interests None.
Ethics approval This study was conducted with the approval of Northwestern University Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves