Article Text

Abstract
Background Long-term benefits of newborn screening (NBS) for cystic fibrosis (CF) have been established with respect to nutritional status, but effects on pulmonary health remain unclear.
Hypothesis With early diagnosis and commencement of standardised treatment, lung function at ∼3 months of age is normal in NBS infants with CF.
Methods Lung clearance index (LCI) and functional residual capacity (FRC) using multiple breath washout (MBW), plethysmographic (pleth) FRC and forced expirations from raised lung volumes were measured in 71 infants with CF (participants in the London CF Collaboration) and 54 contemporaneous healthy controls age ∼3 months.
Results Compared with controls, and after adjustment for body size and age, LCI, FRCMBW and FRCpleth were significantly higher in infants with CF (mean difference (95% CI): 0.5 (0.1 to 0.9), p=0.02; 0.4 (0.1 to 0.7), p=0.02 and 0.9 (0.4 to 1.3), p<0.001, z-scores, respectively), while forced expiratory volume (FEV0.5) and flows (FEF25–75) were significantly lower (−0.9 (−1.3 to −0.6), p<0.001 and −0.7 (−1.1 to −0.2), p=0.004, z-scores, respectively). 21% (15/70) of infants with CF had an elevated LCI (>1.96 z-scores) and 25% (17/68) an abnormally low FEV0.5 (below −1.96 z-scores). While only eight infants with CF had abnormalities of LCI and FEV0.5, using both techniques identified abnormalities in 35% (24/68). Hyperinflation (FRCpleth >1.96 z-scores) was identified in 18% (10/56) of infants with CF and was significantly correlated with diminished FEF25–75 (r=−0.43, p<0.001) but not with LCI or FEV0.5.
Conclusion Despite early diagnosis of CF by NBS and protocol-driven treatment in specialist centres, abnormal lung function, with increased ventilation inhomogeneity and hyperinflation and diminished airway function, is evident in many infants with CF diagnosed through NBS by 3 months of age.
- Infants
- CF
- lung function tests
- newborn infant screening
- cystic fibrosis
- lung physiology
- respiratory measurement
- asthma guidelines
- exhaled airway markers
- paediatric asthma
- paediatric lung disease
- paediatric physician
- paediatric lung disaese
Statistics from Altmetric.com
- Infants
- CF
- lung function tests
- newborn infant screening
- cystic fibrosis
- lung physiology
- respiratory measurement
- asthma guidelines
- exhaled airway markers
- paediatric asthma
- paediatric lung disease
- paediatric physician
- paediatric lung disaese
Key messages
What is the key question?
-
Is lung function normal at 3 months of age in infants with cystic fibrosis (CF) diagnosed via newborn screening?
What is the bottom line?
-
Despite early diagnosis of CF by newborn screening and protocol-driven treatment in specialist centres, abnormal lung function is evident in many screened infants with CF by 3 months of age.
Why read on?
-
This study, the largest of its kind and the only one with contemporaneous healthy controls, describes early lung development in newborn screened infants with CF.
Introduction
The prognosis of cystic fibrosis (CF) has improved dramatically over the years due to implementation of aggressive treatment to optimise nutrition and pulmonary health following diagnosis and increasing global uptake of newborn screening (NBS) for CF.
Despite convincing evidence of the long-term benefits of NBS for CF with respect to improved nutritional status,1 ,2 the extent to which pulmonary outcomes have improved remains controversial. Although some studies have failed to demonstrate any benefits with screening,3 others have reported significantly less pulmonary disease on chest radiography4 and stable lung function (LF) with less marked decline over time in the screened group.5–8 The Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF) group initially reported normal values of forced expired volume in 0.5 s (FEV0.5) in NBS infants with CF in the first 6 months of life,9 whereas a more recent report using slightly different methodology found significant reductions in forced expired flows and volumes during the same period.10
Even in the absence of any clinical respiratory symptoms, infants with CF diagnosed clinically have impaired LF shortly after diagnosis11 ,12 and this impairment persists into school age despite treatment in specialist CF centres.13–15 We have previously shown that the lung clearance index (LCI) measured by multiple breath washout (MBW) is a more sensitive measure of early lung disease in preschool and young school-age children with CF than spirometry,16–18 and that measurements during the preschool years are highly predictive of LF at 6–10 years of age.13 During infancy, complementary information is provided if the forced expiratory manoeuvres19 and MBW are undertaken.20 Nevertheless, the extent to which infant LF tests should be used to help guide CF management during infancy remains controversial.21
Since universal CF NBS was implemented in the UK in October 2007, the median age of CF diagnosis has fallen to ∼1 month.22 However, there have yet to be any large randomised controlled trials of treatment in this age group. For these to be initiated, it is vital to have a greater understanding of the natural history of lung disease in such infants. This study aimed to determine baseline LF within the first 3 months of age in NBS infants with CF and compare findings to prospectively recruited healthy infants of similar age. We hypothesised that, with early diagnosis and commencement of standardised specialist treatment, there would be no difference in LF by 3 months of age in NBS infants with CF compared with healthy babies.
Methods
As part of a longitudinal collaborative research program of infants with CF diagnosed by NBS (see online supplement), this study received ethical approval (#09/HO71/314) from the North Thames Multi-Centre Research Ethics Committee (REC) and the local RECs of the participating specialist centres.
NBS infants with CF born between January 2009 and July 2011 were eligible for recruitment to the London CF Collaboration (LCFC). Healthy full-term infants of similar age were recruited prospectively from a London community (for details, including eligibility criteria, see online supplement, section 2). Lung function tests (LFTs) at 3 months were completed.
Study protocol
Clinical data
With parental consent, participating centres prospectively completed case record forms (CRFs) at diagnosis and each subsequent clinic visit, documenting mode and date of diagnosis, presentation, genotype, history of respiratory symptoms and/or infection, microbiology, treatment, somatic growth and additional investigations undertaken for clinical purposes. These forms enabled auditing and tracking of participating CF centres' adherence to a standardised study treatment protocol (online supplement, section 6) in accordance to the UK CF Trust guidelines.23
Following diagnosis, all infants were commenced on multivitamins, pancreatic supplements if appropriate, and prophylactic flucloxacillin according to the standardised study treatment protocol (online supplement, section 6). Cough swabs were taken every 2–3 months at clinics and whenever symptomatic, using a standardised protocol for collection, storage and analysis of samples.24
Lung function testing and anthropometry
All infants were tested at Great Ormond Street Hospital/UCL Institute of Child Health at ∼3 months postnatal age, when clinically stable and at least 3 weeks after any respiratory tract illness. Infants were weighed and examined, oxygen saturation (SpO2) levels were measured (Masimo Radical-7 pulse oximeter, Irvine, California, USA) and vital signs assessed prior to administering chloral hydrate orally (60–100 mg/kg). Weight and crown-heel length were expressed as z-scores to adjust for age and sex.25 LFTs were performed during epochs of quiet sleep with the child lying supine. Heart rate and SpO2 were monitored continuously throughout testing. Infant urine or maternal saliva samples were collected for cotinine assay to validate parental reported smoking habits (online supplement, section 5, table E4). Parents provided informed written consent and were present throughout measurements.
LCI, a measure of ventilation inhomogeneity, and functional residual capacity (FRCMBW) were measured with MBW, using a respiratory mass spectrometer and customised software.20 The Jaeger BabyBody device (V.4.65; Care Fusion, San Diego, California, USA) was used to measure plethysmographic FRC (FRCpleth),26 ,27 total respiratory compliance (Crs) and resistance (Rrs),28 forced expiratory volume (FEV0.5), forced vital capacity (FVC) and forced expiratory flows (FEF75, FEF25–75) from an inflation pressure of 30 cmH2O using the raised volume technique.19 Measurements were always performed in that order. Details of data collection and analysis as described previously20 ,27–30 are summarised in the online supplement. Results were expressed as z-scores to adjust for body size, sex and age if appropriate using reference equations derived from up to 140 healthy white infants studied in our department over the past decade using identical equipment and protocols19 ,31 ,32 (online supplement, section 3). Abnormal lung function was defined as results outside the 95% limits of normal (ie, below −1.96 z-scores for FEV0.5 or >1.96 z-scores for LCI (<2.5th or >97.5th centile respectively).
Statistical analysis and power of study
Comparisons of group differences were performed using student t tests for continuous variables (age, body size and LFT z-scores) or χ2 analyses for categorical variables (sex, ethnicity, maternal smoking, parental occupation and asthma) (PASW Statistics V.18). Within the CF group, the relationship between LFT outcomes was quantified using Pearson correlations. Multiple linear regression analyses were used to quantify impact of maternal smoking (data not shown) and the extent to which potential clinical determinants (CF genotype, respiratory symptoms, cough swab culture and antibiotic use) are associated with LFT outcomes within the CF group after adjustment for sex and current body size. Model estimates and differences between groups are presented with 95% CIs. Taking into account two primary outcomes (LCI and FEV0.5) a sample size of 70 infants with CF and 50 controls (equivalent to 58 in each group) will allow detection of differences equivalent to 0.58 or 0.66 z-scores at the 5% significance level with 80% or 90% power, respectively.33 Statistical significance was taken as p<0.05.
Results
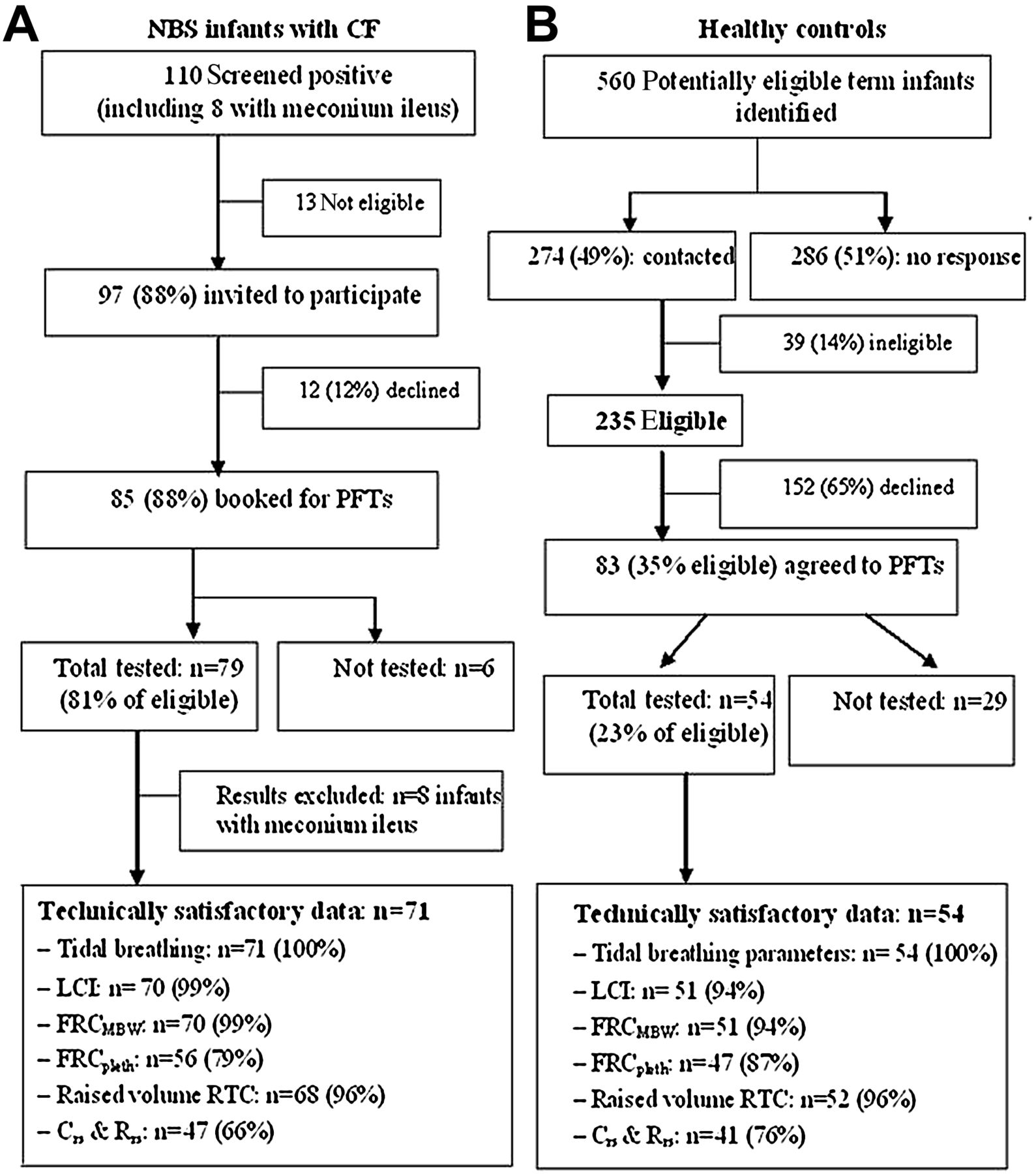
During the study period, 110 infants screened positive for CF, of whom successful LFT measurements were obtained in 79 (81% of those eligible). Inspection of the prospective CRFs, and regular discussion with consultants, suggested that the standardised treatment protocol had been adhered to in all infants at the time of testing. Details are summarised in figure 1, including success rates for each LFT. For clarity, the results presented here are limited to those without meconium ileus (n=71), although including such infants did not affect the results (data not shown). Of the 274 families with potentially eligible healthy infants whom we contacted, 39 (14%) were ineligible. Of the remaining 235, 54 (23% of those eligible) attended for LFTs (figure 1, details in figure E1, online supplement).

Flow diagram showing success rates in relation to recruitment and achievement of technically acceptable infant lung function outcomes in NBS infants with CF (A) and healthy controls (B). CF, cystic fibrosis; Crs, total respiratory compliance; FRC, functional residual capacity; LCI, lung clearance index; MBW, multiple breath washout; NBS, newborn screening; PFT, pulmonary function test; pleth, plethysmographic; Rrs, total respiratory resistance; RTC, Rapid Thoraco-abdominal Compression.
Median (IQR) age at diagnosis for the CF infants was 3.6 (3.1–4.4) weeks; the majority were either homozygous (59%) or heterozygous (32%) ΔF508 and 8 (11%) were pancreatic sufficient. Sixty five (92%) NBS infants with CF had had no respiratory or only mild coryzal symptoms prior to LFTs (figure 2). The remaining 6 (8%) had had significant cough and/or wheeze, 16 (23%) had had at least one positive cough swab by ∼3 months of age (two with Pseudomonas aeruginosa; 10 with Staphylococcus aureus and four with Haemophilus influenzae). These children were prescribed antibiotics according to a standardised treatment protocol (online supplement, section 6) in addition to the prophylactic flucloxacillin prescribed for all infants on diagnosis. No infant had positive cough swab culture for Stenotrophomonos maltophilia; Burkholderia cepacia complex or Aspergillus fumigatus. Additional antibiotics were also prescribed for 36 infants with negative cough swabs, 32 (89%) of whom had had prior cough and/or wheeze. Thus in total, 52 (73%) NBS infants with CF received additional antibiotics prior to their first LFTs at ∼3 months (figure 2), but all were asymptomatic at the time of testing. Of the 46 infants with CF who had cough swabs taken within 10 days of LFTs, only three yielded positive growth (two for S aureus and one for H influenzae).
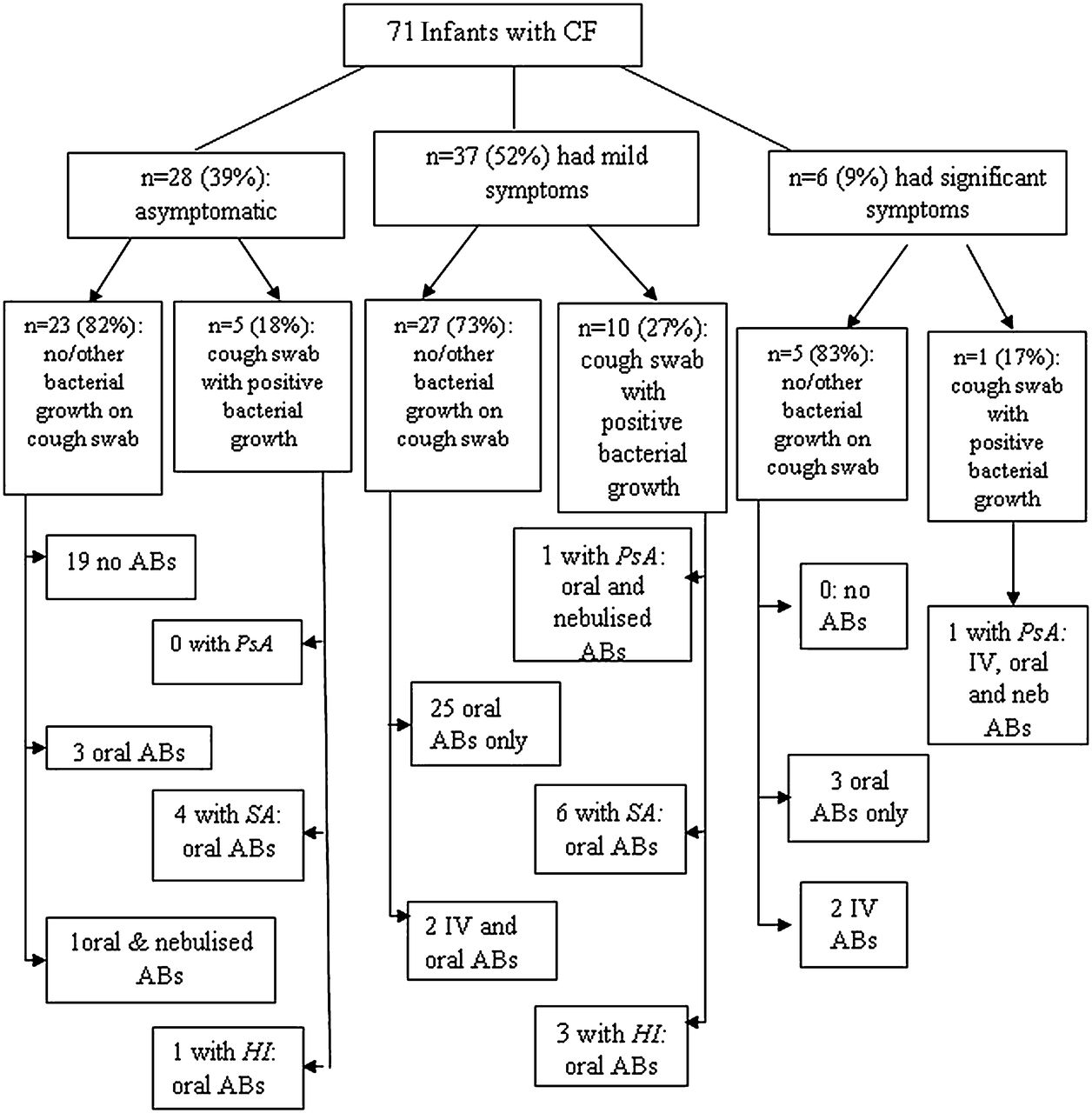
Clinical symptoms and additional antibiotic treatment* of newborn screened infants with cystic fibrosis (CF) prior to lung function tests. Although mothers of infants with CF occasionally reported mild symptoms (slight cough or mild snuffles) in the weeks prior to LFTs, on the day of the test all infants had clear chests on auscultation with no sign of blocked nostrils or cough. Reports of significant symptoms included previous wheeze, crackles, tachypnoea with or without cough and cold. *Antibiotic prescribed in addition to prophylactic flucloxacillin due to respiratory symptoms and/or positive growth on cough swab. See online supplement for standardised treatment protocol. AB, antibiotic; HI, Haemophilus influenzae; IV, intravenous; PsA, Pseudomonas aeruginosa; SA, Staphylococcus aureus.
Table 1 summarises background characteristics of study participants. With the exception of a slightly lower, but statistically significant, gestational age, both groups were very similar.
Comparison of background characteristics in infants with cystic fibrosis* and healthy controls
Infant details at the time of LFTs are shown in table 2. NBS infants with CF were on average 1 week younger than healthy controls, with a significantly greater proportion having birth weights <10th percentile. After taking age and sex into account, infants with CF were significantly lighter and shorter and had a lower body mass index than the controls. The change in weight z-score between birth and LFT was significantly lower in infants with CF than in controls (table 2).
Infant details at time of lung function test
Lung function results
Technically satisfactory measurements of LCI and FRCMBW were obtained in 121 infants (97% of those in whom they were attempted), forced expired volumes and flows in 120 (96%), plethysmographic FRC in 103 (82%), Crs and Rrs in 88 (70%) (figure 1, table 3). Failures were largely due to infants waking prior to completion of the test protocol or technically unacceptable data. With the exception of tidal volume, which was slightly higher (0.4 z-scores) in those with CF, there were no significant differences for any of the tidal breathing outcomes or passive respiratory mechanics (Crs and Rrs). LCI was significantly higher in those with CF, whether expressed as absolute values (online supplement, table E2) or as z-scores (table 3, figure 3). Similarly after adjustment for age, length and sex, FRCMBW was significantly higher (0.38 z-score) in infants with CF. Additional evidence of hyperinflation and gas trapping was seen in those with CF, with FRCpleth and ΔFRC (ie, difference between FRCpleth and FRCMBW z-scores) being significantly elevated (table 3, figure 3). After correction for body size, forced expired flows and volumes were significantly decreased in infants with CF compared with healthy controls, by between 0.5 and 0.9 z-scores, indicating the presence of airways obstruction (table 3, figure 3). The extent to which interpretation of these results would have differed had they simply been expressed as absolute values or weight-corrected ratios is presented in table E2 of the online supplement.
Comparison of lung function in 71 infants with cystic fibrosis and 54 healthy controls

{kind=link}
{kind=link}
{kind=link}
Comparison of lung function outcomes between infants with cystic fibrosis and healthy controls. As expected, approximately 95% of healthy controls had values within ±1.96 z-scores, with none having an abnormal FEV0.5 z-score, 2/51 (4%) having an elevated LCI z-score, and 2/47 (4%) an elevated FRCpleth z-score. FEF25-75, forced expiratory flow between 25% and 75% of vital capacity; FEV0.5, forced expiratory volume in 0.5 s; FRCpleth, plethysmographic functional residual capacity; LCI, lung clearance index; MBW, multiple breath washout; Rrs, total respiratory resistance.
Additional determinants of lung function
On multivariable analyses, after adjustment for CF, other potential determinants (sex, gestational age, birth weight z-score, pre- or postnatal maternal smoking and maternal asthma) were not significantly associated with any LF z-scores. Among infants with CF, with the exception of a significantly lower FEV0.5 (mean (95% CI): −0.70 (−1.29 to −0.10) z-scores) in those who had received any additional antibiotics for symptoms or positive cough swab, there was no significant association between LF outcomes and the infants' genotype, clinical status or treatment prior to LFTs at ∼3 months of age (see table E3, OLS).
Relationship between different lung function outcomes in infants with CF
The relationship between selected LF outcomes in infants with CF is shown in figure E2 (online supplement). There was no significant relationship between the two primary outcomes FEV0.5 and LCI (r=−0.10, p=0.432). Twenty-one percent of infants had an LCI above 1.96 z-scores, whereas 25% had an FEV0.5 below −1.96 z-scores, while only 12% (8/68) had abnormalities detected by both these tests (figure E2a); if based on either test, 35% (24/68) would be identified with abnormal results. FEF25–75 and FEV0.5 (r=0.73, p<0.001) detected a similar proportion of infants outside the normal range (24% and 25%, respectively, figure E2b), whereas FEF75 was less discriminative (only detecting abnormalities in 15% of infants; data not shown). FRCpleth z-score and ΔFRC were highly correlated (r=0.66, p<0.001, figure E2d), both detecting a similar proportion of infants with abnormally elevated results (18% and 20%, respectively). Forty-four percent (31/71) of NBS infants with CF had at least one abnormal result if based on LCI, FEV0.5 or FRCpleth.
Discussion
In this, the largest study of its kind and the only one with contemporaneous healthy controls, we have shown that, by 3 months of age, many NBS infants with CF have reduced forced expired flows and volumes, abnormal gas mixing and hyperinflation, despite early diagnosis and protocol-driven management, which included prophylactic oral flucloxacillin from time of diagnosis.
Strengths and weaknesses
The major strengths of this study include the fact that all lung function measurements were performed to international standards in a single centre by a highly experienced team, thereby minimising methodological or analytical bias. Accurate identification of the extent to which abnormalities in LF were present in individual infants was facilitated by expressing results as z-scores. These were derived from equipment-specific reference equations based on a large group of healthy infants studied in our department with the same equipment and methods over the past decade.31 ,32 The ability to recruit a large group of contemporaneous healthy controls specifically for this study further strengthened the confidence with which we could detect changes due to CF lung disease after adjusting for other relevant determinants such as body size, age and sex.34 ,35
Inevitably, it was necessary to approach a large number of families of healthy infants to recruit appropriate numbers for comparison, with only 25% of those eligible and whom we could contact actually attending for the tests. This could potentially introduce some bias, especially as due to data protection issues, it was not possible to document background data from those not responding or agreeing to participate. However, it was reassuring to find no significant differences with respect to potential determinants of infant LF such as ethnicity, maternal history of asthma, socioeconomic status (based on parental occupation) or exposure to tobacco smoke between the controls and infants with CF, suggesting that the group were representative of the local population. Furthermore, when expressed as z-scores, both anthropometry and LF were very similar in this current healthy control group as in those previously recruited in London by our team.15 ,20 Travel expenses were reimbursed, but no incentives were provided to encourage attendance for either group, thereby removing a potential source of bias. Infants with CF were, on average, born 1 week earlier than the controls, a pattern that we have noted in previous studies.11 Any potential group differences due to developmental changes were avoided by studying all infants within a narrow age range; mean difference at time of test being less than a week.
Of the 110 infants screening positive for CF over the 2.5-year recruitment period, 12% were excluded due to coexistent morbidity or psychosocial reasons, with tests not possible in a further five infants before 4 months of age due to recent exacerbations and deferment of appointments. If anything, the results from this study therefore underestimate the degree of morbidity by 3 months of age. Nevertheless, with only 13% of parents declining participation (including one family that withdrew after initial consent), we were able to test 79/110 (72%) of the entire cohort and 81% of those eligible.
Since lung function was assessed in the first few months of life, we excluded results from those presenting with meconium ileus to preclude any adverse effects related to surgery, although subsequent sub-analysis indicated that the results were not affected by including such children. All infants were tested at least 3 weeks after a respiratory illness when free of symptoms, in an attempt to minimise impact of acute exacerbations. This did mean that we had to exclude several healthy controls in whom it was impossible to rearrange appointments within the designated age range for this study of ‘early LFT’ (figure E1, online supplement). While it could be argued that the observed differences in LF between CF and controls may simply represent post-respiratory tract infection changes in those with CF, this is unlikely given the lack of association between LF and respiratory symptoms in those with CF, except for a significant reduction in z-FEV0.5 when infants had received additional antibiotics (table E3, online supplement).
To optimise recruitment to this observational study and to ensure initial LFTs could be undertaken within the first months of life, routine bronchoscopy and bronchoalveolar lavage were not included within this study protocol but, together with chest high-resolution CT scans, will be included when these children are reassessed at 1 year of age.
A further strength of this study was the wide range of tests applied, minimising the chance that early changes in LF would be missed. As in a previous study of infants,20 but in contrast to findings in older children,13 ,16 ,17 ,36 we found that assessments of lung disease based on forced expiratory manoeuvres and ventilation inhomogeneity were necessary to identify early lung disease during the first year of life, with additional useful information regarding hyperinflation being obtained from plethysmographic lung volumes. The slightly lower absolute mean LCI observed in infants with CF in this study (table E2, online supplement) than in our previous study20 probably reflects the fact that these infants were diagnosed by NBS rather than clinically, and were therefore assessed earlier in the disease process. Had assessments been limited to a single technique, abnormalities would only have been detected in ∼25% of infants with CF, this proportion rising to 35% when using either of the two primary outcomes (LCI and FEV0.5). The relatively poor correlation between these various outcomes suggests that they are reflecting different aspects of underlying pathophysiology. Despite their relative simplicity, assessments of tidal breathing variables and passive mechanics were far less discriminatory than the other techniques and could be usefully omitted when investigating infants with CF.37 While Crs results were lower and Rrs higher in those with CF when expressed as absolute values (table E2, online supplement), this was largely related to body size at the time of testing; Crs increasing and Rrs decreasing with somatic growth.32 ,38 In contrast to all other outcome measures investigated, after adjusting for length and/or age, no significant differences were observed with respect to either Crs or Rrs (table 3).
Clinical status
Despite early diagnosis and commencement of pancreatic enzyme replacement therapy, vitamin supplement and prophylactic antibiotics, infants with CF experienced significantly slower growth during the first few months and were significantly lighter and shorter than their healthy peers by the time of the 3-month LFTs. Furthermore, by this age, 61% of the screened infants had had some respiratory symptoms (52% mild, 9% severe), 23% a positive cough swab and 73% had received antibiotics in addition to their routine prophylactic medication. Pulmonary involvement is known to be present early, with some infants with CF having evidence of inflammation in the bronchoalveolar lavage fluid as early as 4 weeks of age.39–41 With the exception of a significantly lower FEV0.5 in those who had received additional antibiotics for symptoms or positive cough swab, there was no significant association between LF outcomes and the infant's genotype, clinical status, growth trajectory or treatment prior to the LFTs at 3 months of age. Of note, many infants who had been treated aggressively for respiratory exacerbations in the first few months had entirely normal LF by 3 months, whereas others with no prior symptoms or cause for concern had evidence of early lung disease.
Comparison with the literature
The commonest characteristics of LF abnormalities described in CF lung disease during the first years of life have been airway obstruction detected using the raised volume technique,10–12 ,15 ,20 hyperinflation indicated by elevated resting lung volumes,42 increased ventilation inhomogeneity20 and, in infants and slightly older children, gas trapping,17 ,18 ,43 ,44 all of which were observed in this study by 3 months of age in infants diagnosed by NBS.
In a US multicentre evaluation of LF in infants with CF aged 4–24 months (21% diagnosed by screening),42 elevated lung volumes and diminished forced expiratory flows, but no reduction in FEV0.5, were reported when compared with historical controls. The variability in skill mix and experience of the laboratories participating in that study, together with the lack of contemporaneous healthy controls and different age range studied, may have contributed to differences in findings compared with the current results.35
The Australian AREST-CF study recently published LF results from infants with CF diagnosed by NBS.9 ,10 When using a lung inflation pressure of 20 cmH2O during forced expiratory manoeuvres, LF was reported to be normal during the first 6 months of life, but thereafter declined at a rapid rate.9 However, the number of infants studied during the first months of life was limited. By contrast, a more recent publication from this group,9 ,10 reporting measurements from 28 NBS infants with CF within the first 6 months of life when using an inflation pressure of 30 cmH2O as recommended by the American Thoracic Society/European Respiratory Society guidelines19 showed diminished LF at the time of the first test, with continued deterioration over the next 2 years of life when the results were compared with published reference equations.45
Clinical implications
The results from this study indicate that despite early diagnosis and rapid implementation of therapy, including prophylactic antibiotics, a substantial number of NBS infants with CF have abnormalities of LF within the first 3 months of life. The apparent wellness of the cohort should not lead to complacency, and prompt and aggressive treatment of any abnormal symptoms or signs is surely vital. Follow-up of this cohort will be essential to ascertain the extent to which early changes in LF persist throughout the first year of life; if there is catch-up growth with conventional treatment, then novel, molecular-based therapies46 ,47 may be safely deferred; if not, there will be a compelling case for initiating treatment early in these infants, using physiological endpoints to detect benefit.
Conclusions
Despite early diagnosis of CF by NBS and protocol-driven treatment in specialist centres, abnormal LF, with increased ventilation inhomogeneity and hyperinflation and diminished airway function, is evident in many infants with CF diagnosed through NBS by 3 months of age. CF clinicians should not be lulled into thinking that babies with CF identified by NBS have good pulmonary health in the first few months of life.
Acknowledgments
We thank the infants and parents who participated in this study and contributions by members of the London CF Collaboration (including Janet Stocks, Andy Bush, John Price, Colin Wallis, Ranjan Suri, Paul Aurora, Ian Balfour-Lynn, Siobhan Carr, Caroline Pao, Hilary Wyatt, Gary Ruiz, Richard Chavesse, Anu Shanker, Wanda Kozlowska, Ah-Fong Hoo, Sooky Lum, The Thanh Diem Nguyen, Lena Thia, Jane Chudleigh, and Ammani Prasad). We also thank Per Gustafsson for advice and assistance regarding the multiple breath inert gas washout technique.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement 1
- Data supplement 2 - Online supplement 2
Footnotes
Linked article Yes.
Funding This study is supported by grants from the Cystic Fibrosis Trust, UK; Special Trustees: Great Ormond Street Hospital for Children, London, UK; Smiths Medical Ltd, UK; Comprehensive Local Research Network, UK. It was also supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was granted by the North Thames Multi-centre Research Ethics Committee (REC) (#09/HO71/314) and local REC of the participating specialist centres.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves