Article Text

Abstract
Background Fluticasone furoate (FF) is a novel inhaled corticosteroid with 24 h activity. FF is being developed as a once-daily treatment in combination with the long-acting β2 agonist vilanterol trifenatate for asthma and chronic obstructive pulmonary disease.
Objectives To determine the optimal dose(s) of FF for treating patients with asthma.
Methods An 8-week multicentre, randomised, double-blind study. 627 patients with persistent moderate-to-severe asthma, symptomatic on medium-dose inhaled corticosteroid therapy, were randomised to placebo, FF 200, 400, 600 or 800 μg (once daily in the evening using a novel dry powder inhaler), or fluticasone propionate 500 μg twice daily (via Diskus™/Accuhaler™). The primary efficacy measure was mean change from baseline in pre-dose evening forced expiratory volume in one second (FEV1). Other endpoints included morning and evening peak expiratory flow, and rescue/symptom-free 24 h periods.
Results Each dose was significantly superior to placebo for the primary endpoint (p<0.001) with efficacy at least similar to that reported with fluticasone propionate. There was no dose–response relationship across the FF doses studied. Peak expiratory flow improved in all groups (p<0.001 vs placebo), and there were significant treatment effects on rescue/symptom-free 24 h periods with all active treatments. FF was generally well tolerated. The incidence of oral candidiasis was higher with FF 800 μg than placebo; pharmacokinetic and 24 h urinary cortisol analyses confirmed a higher systemic exposure of FF at this highest dose level.
Conclusions FF doses <800 μg have a favourable therapeutic index. The absence of an efficacy dose response suggests that 200 μg is an appropriate dose in patients with moderate persistent asthma.
ClinicalTrials.gov identifier NCT00603746.
- Asthma
- dose–response
- evening dosing
- fluticasone furoate
- fluticasone propionate
- inhaled corticosteroids
- once-daily dosing
- asthma epidemiology
- asthma genetics
- asthma pharmacology
- drug reactions
- asthma guidelines
- asthma in primary care
- COPD mechanisms
- COPD epidemiology
- inhaler devices
- COPD pathology
- eosinophil biology
- exhaled airway markers
- allergic lung disease
- cough/mechanisms/pharmacology
Statistics from Altmetric.com
- Asthma
- dose–response
- evening dosing
- fluticasone furoate
- fluticasone propionate
- inhaled corticosteroids
- once-daily dosing
- asthma epidemiology
- asthma genetics
- asthma pharmacology
- drug reactions
- asthma guidelines
- asthma in primary care
- COPD mechanisms
- COPD epidemiology
- inhaler devices
- COPD pathology
- eosinophil biology
- exhaled airway markers
- allergic lung disease
- cough/mechanisms/pharmacology
Key messages
What are the key questions?
Can the once-daily inhaled corticosteroid fluticasone furoate (200–800 μg) provide effective asthma control with an acceptable tolerability profile?
What is the bottom line?
Fluticasone furoate <800 μg once daily provided clinically and statistically relevant improvements in lung function and symptom control, and was well tolerated in adult patients with moderate persistent asthma.
Why read on?
Poor adherence to twice-daily therapy in asthma is associated with poor asthma control; once-daily dosing with fluticasone furoate (<800 μg) in asthma may improve treatment compliance and provide effective asthma control with good tolerability.
Introduction
Asthma treatment is designed to achieve long-term control of symptoms and lung function and to reduce future risk of acute exacerbations and mortality.1 2 Despite the availability of effective treatments and established guidelines, poor asthma control contributes to the significant socioeconomic and healthcare burden associated with this disease.3 4 Long-term inhaled corticosteroid (ICS) therapy is considered the most effective anti-inflammatory treatment for all severities of persistent asthma.1 3 5 Treatment adherence can be low6 7 and contributes to poor asthma control. Fluticasone furoate (FF) is a novel ICS with persistent effects at 24 h because of enhanced affinity in the lung and it has a longer duration of action than fluticasone propionate (FP) as demonstrated in preclinical and early clinical studies.8 9 The structures of FF and FP are different; FF has an ester derived from 2-furoic acid at the C-17a position replacing the simpler propionate ester found in FP, resulting in more complete occupancy of the 17a pocket in the glucocorticoid receptor.10 This difference results in higher glucocorticoid receptor binding affinity with FF.8
FF is currently under development for use as a once-daily inhaled treatment for asthma and chronic obstructive pulmonary disease, both alone (asthma) and in a fixed-dose combination with the novel long-acting β2 agonist (LABA), vilanterol trifenatate (GW642444M). FF is administered using a novel dry powder inhaler (DPI).
Results from a previous phase II dose-ranging study of inhaled FF found 100 μg, given once daily in the evening, was optimal for controlling asthma in patients who were symptomatic despite receiving non-steroidal therapy.11 The present study was conducted to evaluate the efficacy and safety of higher doses of FF (200, 400, 600 and 800 μg) in patients with moderate persistent asthma who remain symptomatic on medium-dose ICS therapy. Preliminary results from this study have previously been presented in abstract form.12
Patients and methods
Patients and screening
Patients aged 12 years or older were eligible if they had persistent asthma that was not controlled using medium-dose ICS (FP 500 μg/day or equivalent and stable at this dose for the last 4 weeks). Patients had asthma according to National Institutes of Health criteria.1 Eligible patients demonstrated reversibility of forced expiratory volume in 1 s (FEV1) of at least 12% and 200 ml with inhaled salbutamol and an FEV1 of 40–90% of predicted normal between 17:00 and 23:00 (or 40–85% between 05:00 and 12:00).1 National Health and Nutrition Examination Survey III values were calculated for all patients. Exclusion criteria and prohibited medications are shown in the online supplement.
Electronic case report forms were provided to record patient data. A 4-week pre-treatment run-in period enabled baseline assessment of symptoms and safety measures; patients continued their current ICS therapy at regular fixed doses with salbutamol as needed. At the end of the run-in period, eligibility was confirmed if patients were compliant with treatment and completion of a daily diary; evening pre-dose FEV1 was 40–90%; and symptoms continued (combined daytime and night-time symptom score ≥1 or use of as needed salbutamol on at least 4 of the last 7 days).
Study design and treatments
This phase IIb study (FFA109684; ClinicalTrials.gov identifier NCT00603746) was conducted in 94 centres in 16 countries worldwide between 20 December 2007 and 20 September 2008 in accordance with Good Clinical Practice guidelines, all applicable regulatory requirements and the Declaration of Helsinki. The study protocol and consent process were reviewed and approved by a local ethics committee or institutional review board and written informed consent was obtained from each patient. The study had a randomised, double-blind, double-dummy, parallel-group, placebo-controlled design to evaluate the efficacy and safety of four once-daily doses of FF. A separate FP group was included as an active control of assay sensitivity.
Following the 4-week run-in period, eligible patients were randomised (approximately 1:1) to receive one of the following: one of four doses of FF (200 μg, 400 μg, 600 μg or 800 μg) once daily (evening) via a novel single-step activation DPI or placebo twice daily (morning and evening) via Diskus™/Accuhaler™; FP 500 μg twice daily (morning and evening) via Diskus/Accuhaler plus placebo once daily (evening) via novel DPI; or placebo once daily (evening) via novel DPI and placebo twice daily (morning and evening) via Diskus/Accuhaler. Patients and investigators were blinded to treatment assignment; the placebo and FF formulations were indistinguishable. Details of blinding and randomisation schedule (RandAII) and allocation method (Registration and Medication Ordering System (RAMOS)) are described in the online supplement.
Treatment was continued for 8 weeks, with assessments after 1, 2, 4, 6 and 8 weeks, with a follow-up 1 week after completion of the trial. Appropriate device use and adherence to study treatment (using the device dose counters) were assessed at each visit.
Patients stopped their usual ICS medication at randomisation and, while on study treatment, were not permitted to use any other asthma medication other than the salbutamol inhaler provided. Any other medications taken were recorded in the electronic case report form; stable-dose immunotherapy and intranasal or topical corticosteroids were permitted.
Assessments
The primary endpoint was mean change from baseline in trough evening FEV1 (measured at evening clinic visits pre-dose and pre-rescue bronchodilator) at the end of the 8-week treatment period. Although not a primary endpoint, changes in FEV1 at weeks 1, 2, 4 and 6 were also analysed.
Efficacy measures for secondary endpoints recorded by patients using the daily diary were combined daytime and night-time asthma symptom scores for each 24 h period, rescue salbutamol use and morning and evening peak expiratory flow (PEF) measurements (pre-dose, pre-rescue bronchodilator). Other efficacy endpoints included proportion of symptom-free days, rescue-free days and withdrawals due to worsening asthma.
Adverse events (defined using the Medical Dictionary for Regulatory Activities V.11) were documented during the 8-week treatment period. Asthma exacerbations, defined as the need for systemic corticosteroids, or emergency room visit or hospitalisation due to asthma requiring systemic corticosteriods, were also recorded. Standard laboratory parameters and 24 h urinary cortisol excretion were measured before and at the end of treatment. Vital signs and oropharyngeal examination for signs of oral candidiasis were assessed at screening and all on-treatment visits.
Blood samples for pharmacokinetic assessment of FF were collected from all patients pre-dose and 30–120 min post-dose at weeks 2 and 8 of study treatment. Samples from patients receiving FF were analysed using validated methods based on solid phase extraction followed by high-performance liquid chromatography, with a lower limit of quantification of 10 pg/ml.
Analysis
Approximately 1200 patients were screened to identify 594 evaluable patients (99 per treatment group), providing 96% power to demonstrate a 200 ml improvement per 800 μg of FF (dose–response slope of 0.25 ml/μg) in the primary endpoint (change from baseline in trough FEV1), with significance at the two-sided 5% level. Additionally, the study had 90% power to detect a 200 ml difference in pair-wise comparisons between any active dose and placebo.
All patients randomised to treatment and who received at least one dose of study medication were included in the intent-to-treat (ITT) population. Patients without full protocol deviations were included in the per protocol (PP) population. The urinary cortisol population consisted of patients whose urine samples did not have confounding factors. Patients receiving FF for whom a pharmacokinetic sample was obtained and analysed constituted the pharmacokinetic population.
All statistical analysis was performed using SAS V.9. The primary endpoint analysis was a test for linear dose–response trend in trough evening FEV1 at week 8 across the four doses of FF and placebo, using analysis of covariance with last observation carried forward to impute missing data. If the trend was significant, each dose of FF and FP was compared pair-wise with placebo to identify effective doses. For the secondary treatment comparisons, the linear dose–response test was repeated excluding placebo, and pair-wise comparisons were performed for the other key efficacy endpoints and for 24 h urinary cortisol excretion.
Population pharmacokinetic analysis, using non-linear mixed effect modelling, was performed with the web-based computer program NONMEM, V.V. The effects of patient demographic characteristics such as gender, age, weight, body mass index and ethnicity were also examined.
Results
Study population
Of 1175 patients screened, 627 were randomised, 622 received at least one dose of study treatment (ITT population) and 540 did not have any full protocol deviations (PP population) (figure 1). Patients' demographic characteristics, lung function and run-in mediation use were evenly matched across study groups (table 1). Mean FEV1 percentage predicted at screening was 64.12–66.59%. Between 80% and 90% of patients had suffered from asthma for at least 5 years, and less than 20% experienced an exacerbation during the preceding 6 months.

Patient disposition (CONSORT). *n=1, †n=3, ‡n=1 patient(s) who failed screening but were still randomised. BD, twice daily; FF, fluticasone furoate; FP, fluticasone propionate; ITT, intent-to-treat; OD, once daily; PK, pharmacokinetic.
Demographics (intent-to-treat population)
Efficacy
Lung function
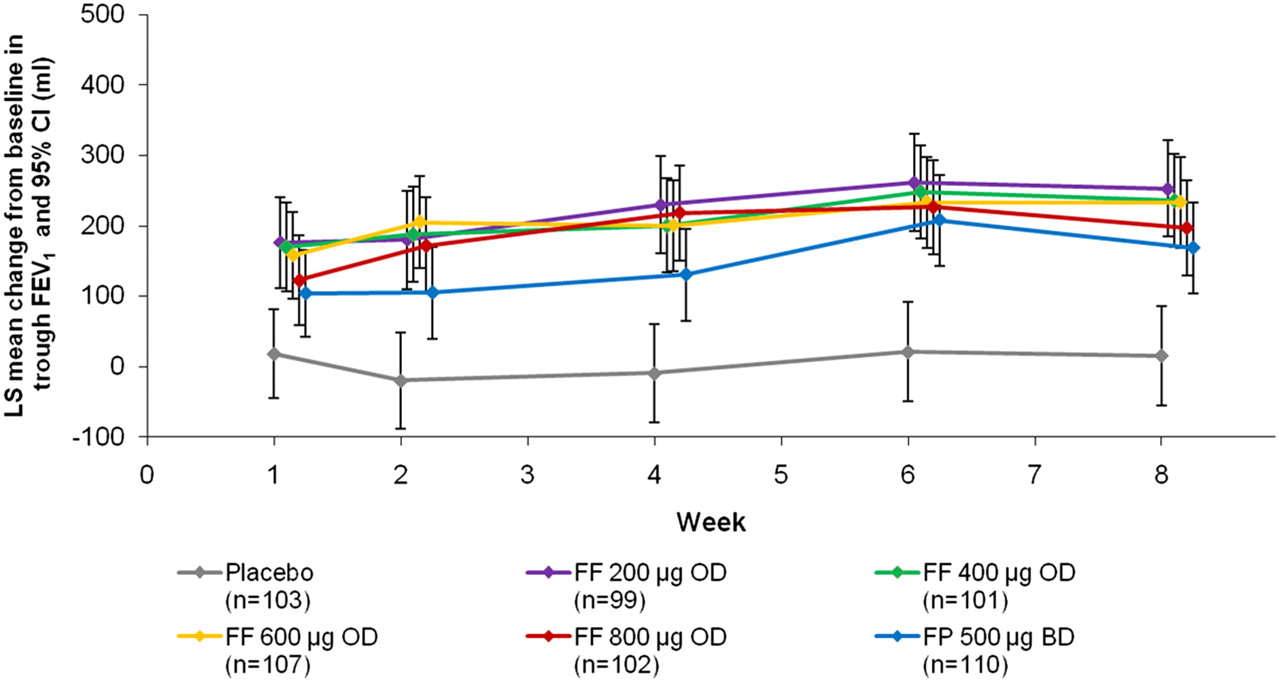
A statistically significant linear trend test (p<0.001) was observed, with a non-zero slope for trough FEV1 against FF dose indicating overall efficacy of FF relative to placebo. However, when placebo was excluded from the analysis, no significant linear trend (p=0.306) was observed. The primary endpoint of the study was mean change from baseline in trough FEV1 at the end of the 8-week treatment period, and the overall efficacy of FF versus placebo allowed pair-wise comparisons of each dose with placebo. Superiority versus placebo was confirmed for each FF dose and for FP, in terms of evening trough FEV1 at week 8 (p<0.001). The predefined 200 ml difference relative to placebo was achieved in all FF groups (figure 2). Results in the PP population were consistent with those described for the ITT population (data not shown). The effect of FF was evident by week 1, increased until week 3, and was sustained until the end of the study (figure 3).

Change from baseline in trough evening FEV1 at week 8 (intent-to-treat population; last observation carried forward). Values are least squares difference from placebo in ml. Error bars indicate 95% CI. BD, twice daily; FEV1, forced expiratory volume in 1 s; FF, fluticasone furoate; FP, fluticasone propionate; OD, once daily.

Least squares mean change from baseline in trough FEV1. Repeated measures analysis (intent-to-treat population). Error bars indicate 95% CI. Data points are offset for clarity. BD, twice daily; FEV1, forced expiratory volume in 1 s; FF, fluticasone furoate; FP, fluticasone propionate; LS, least squares; OD, once daily; PBO, placebo.
Improvements from baseline in evening PEF over the treatment period were significantly greater than placebo in all active treatment groups (p<0.001); patients in the placebo group showed a deterioration in PEF (–5.1 litre/min). The greatest mean increases were observed in the FF 400 μg and 800 μg groups (14.5 and 16.3 litre/min, respectively, compared with 11.1 to 11.9 litre/min for the other FF groups and the FP group). Figure 4 shows the mean change from baseline in evening PEF over time. Similar improvements from baseline were also observed in morning PEF over the 8-week treatment period (p<0.001 for all FF and FP groups vs placebo).

Mean change from baseline in evening PEF (litre/min) (intent-to-treat population). BD, twice daily; FF, fluticasone furoate; FP, fluticasone propionate; OD, once daily; PEF, peak expiratory flow.
Other endpoints
Based on patient-reported data, the proportion of symptom-free 24 h periods during weeks 1–8 increased relative to baseline in all study groups and was greater with all FF doses and FP than placebo (table 2). Similar results were observed for rescue-free 24 h periods (table 2). The proportion of patients with symptom-free days and rescue-free days were also significantly greater in the FF and FP groups than in the placebo group (comparisons vs placebo p<0.001, except for p=0.006 with FF 600 μg for symptom-free days).
Symptom-free and rescue-free 24 h periods over weeks 1–8 (intent-to-treat population)
Withdrawal rates due to lack of efficacy (figure 5) were significantly lower in all active treatment groups versus placebo (6–12% vs 33%; p<0.001 for all comparisons). The fewest withdrawals due to lack of efficacy occurred in the FF 400 μg and FP groups (6% and 7%, respectively).

Time to withdrawals due to lack of efficacy (cumulative incidence curve) (intent-to-treat population). BD, twice daily; FF, fluticasone furoate; FP, fluticasone propionate; OD, once daily.
Pharmacokinetics and pharmacodynamics
A total of 1408 samples from 368 patients receiving FF were included in the pharmacokinetic analysis. FF plasma concentration–time data were well described by a one-compartment model with first-order absorption and elimination and moderate to high between-subject variability. The fraction of dose absorbed could not be estimated and was considered to remain constant over the dosing period. Area under the plasma concentration–time curve over the dosing interval (AUC(0–24)) was 525, 938, 1396 and 1776 pg.h/ml for the 200, 400, 600 and 800 μg doses respectively. FF clearance was estimated at 530 litre/h with between-subject variability of 48%; body mass index was the only significant covariate on this parameter.
Safety
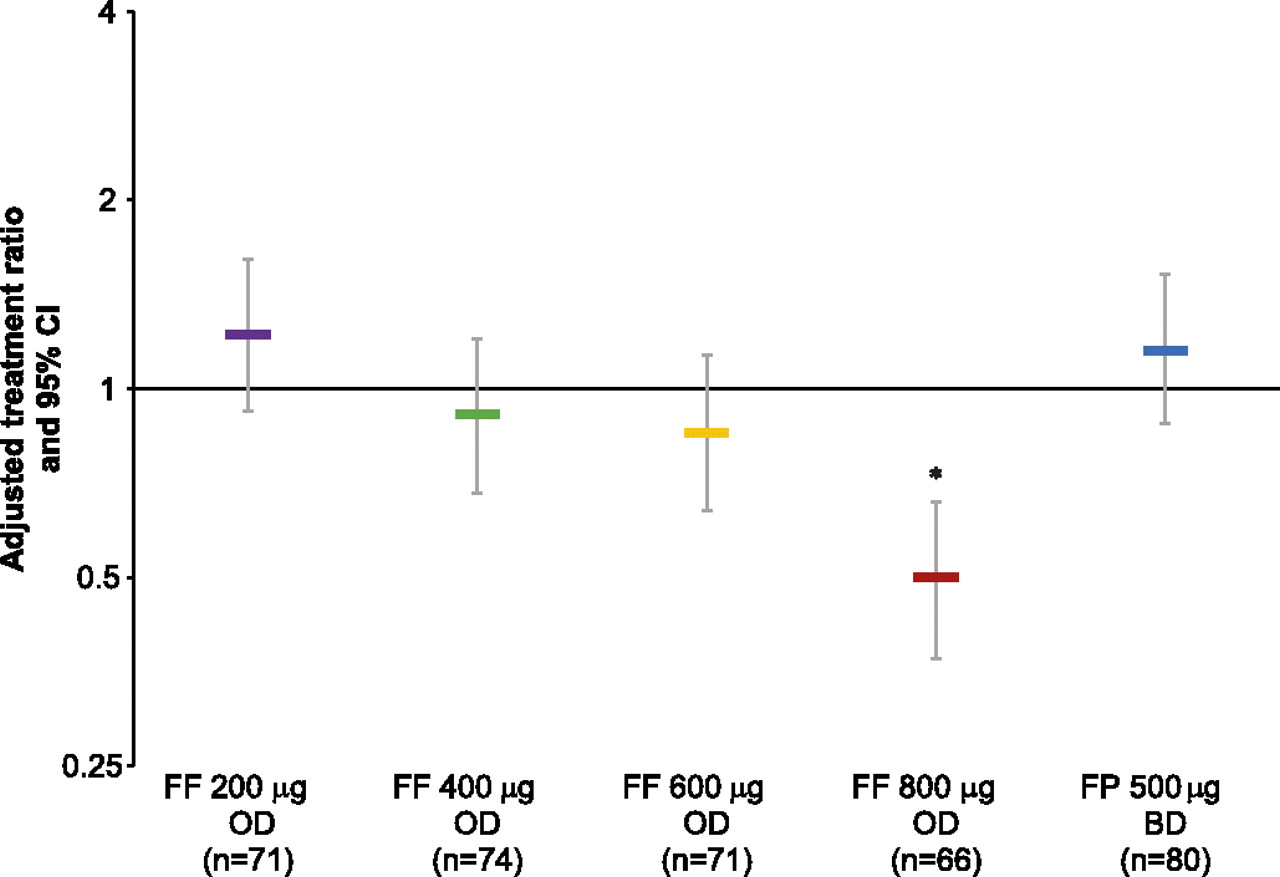
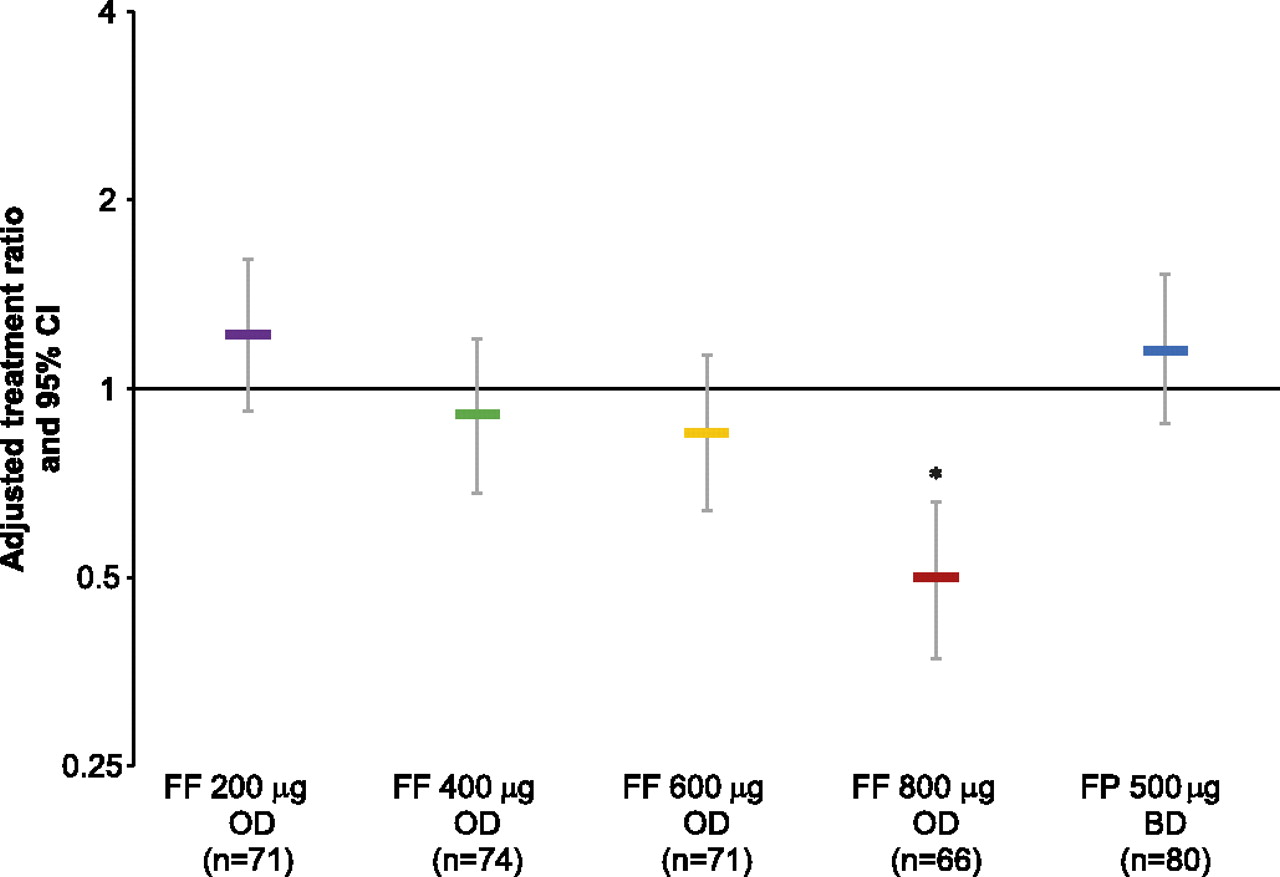
Mean exposure to study medication was similar across the active treatment groups (50–54 days) and somewhat lower for placebo (43 days); adherence to study medication was consistently high across study groups (93.4–96.0% for Diskus/Accuhaler; 98.5–99.7% for novel DPI). For 24 h urinary cortisol excretion (week 8 vs baseline ratio), the FF 800 μg group was significantly different from placebo (FF to placebo ratio 0.5 (p<0.001); figure 6). None of the other FF groups showed statistically significant differences from placebo, with ratios to placebo of 0.85–1.22.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adjusted treatment ratios for 24 h urinary cortisol excretion (urinary cortisol population). Error bars indicate 95% CI. *p<0.001 for FF 800 μg versus placebo at week 8. BD, twice daily; FF, fluticasone furoate; FP, fluticasone propionate; OD, once daily.
The incidence of asthma exacerbations was lower in the active treatment groups (FF 200 μg, n=6 (6%); FF 400 μg, n=1 (<1%); FF 600 μg, n=4 (4%); FF 800 μg, n=1 (<1%); FP 500 μg twice daily, n=6 (5%)) than in the placebo group (n=16 (16%)). Most exacerbations in the placebo group were attributed to lack of efficacy. Eight per cent of patients in the placebo arm required oral corticosteroids compared with 0–2% in the FF groups and 3% in the FP group. Three patients were hospitalised due to asthma exacerbation, one each in the placebo, FF 200 μg once daily and FP 500 μg twice daily arms.
Overall, FF was well tolerated (table 3); 31–35% of patients in the FF groups and 22% in the placebo group experienced one or more adverse event during treatment. The most frequently reported adverse events were oral candidiasis reported as ‘oropharyngeal candidiasis’, ‘oral candidiasis’ or ‘candidiasis’ (<1–12% across treatment groups), headache (3–11% across treatment groups), nasopharyngitis (2–7%) and dysphonia (<1–5%). The incidence of drug-related adverse events was 2% in the placebo group and 11%, 11%, 3%, 17% and 9% of patients in the FF 200, 400, 600 and 800 μg groups and FP group, respectively; the most frequent of these were oropharyngeal candidiasis, oral candidiasis and dysphonia. The frequency of these events was similar in all active treatment groups, with the exception of oral candidiasis, which occurred most frequently in the FF 800 μg group. No safety concerns were noted from the results of vital sign assessments or laboratory safety tests.
Most common on-treatment adverse events (occurring in 3% or more patients in any treatment group) (intent-to-treat population)
No deaths occurred during the study. Nine serious adverse events occurred in six patients (0–2% of patients in each group); none were considered related to study treatment. Five of these patients withdrew from the study: one in the placebo group (asthma exacerbation), two in the FF 200 μg group (acute intestinal infection; pneumonia), and two in the FP group (amitriptyline overdose; asthma exacerbation). Five further withdrawals were linked with non-serious adverse events: one in each of the placebo, FF 200 μg and FF 600 μg groups, and two in the FP group.
Discussion
Eight weeks' treatment with inhaled FF (200–800 μg), given once daily in the evening with a novel single-step activation DPI, produced effective asthma control in patients with moderate persistent asthma previously uncontrolled on medium doses of ICS. The primary analysis showed a significant overall improvement in lung function compared with placebo (evening trough FEV1 at week 8 vs baseline; p<0.001) and all FF dosages individually demonstrated superior improvement in evening trough FEV1 from baseline relative to placebo (p<0.001). Although not directly compared with once-daily FF, the twice-daily FP study arm also provided an appropriate control; however, the improvement in weekly trough FEV1 was numerically lower with twice-daily FP (figure 3).
The results of a similar dose-ranging study of FF at lower doses (25–200 μg, once daily) in patients with non-severe, persistent asthma (uncontrolled on non-steroidal asthma therapy but not receiving regular ICS), demonstrated a significant, dose-dependent effect on trough evening FEV1 and significant improvement over placebo at dose levels ≥50 μg.11 A dose-dependent effect on FEV1 was not observed across the higher FF dosages in the present study, however the higher doses investigated may be near or at the top of the dose–response curve for the FEV1 endpoint in patients with moderate persistent asthma. The trough FEV1 primary endpoint was selected in this study as a reliable and reproducible measure of 24 h improvement in airflow observation.3 Other endpoints including exacerbation frequency and asthma control (eg, the extent of rescue-free and symptom-free 24 h periods assessed as secondary endpoints in this study), and laboratory measures of inflammation (such as sputum eosinophil counts) in addition to FEV1 may be useful in assessing the potential dose-related benefits of FF as these endpoints may have greater sensitivity at higher doses.
Secondary analyses of lung function (evening and morning PEF improved in all groups; p<0.001 vs placebo) and symptom control (significant treatment effects on rescue-free and symptom-free 24 h periods with all active treatments) confirmed efficacy in all active treatment groups. Consistent with the primary endpoint, no dose–response relationship was evident. Although the study was not powered to detect differences between active treatment groups, all once-daily FF dosages produced efficacy at least similar to that seen with twice-daily FP.
The results of both the lung function and symptom control endpoints in this study provide further evidence for the 24 h duration of effect of once-daily FF.8 9 These data demonstrate the feasibility of FF as the 24 h ICS component of a once-daily combination treatment for asthma control. The combination of FF with vilanterol trifenatate, a once-daily LABA with inherent 24 h activity,13 14 is under study. The once-daily approach may improve adherence to asthma control therapy,15 a significant factor in the poor control and high burden of the disease.3 7 16 17 FF was administered in the evening, in accordance with growing acceptance18 that this timing achieves better asthma control than morning dosing with ICS.19–21
Lower rates of withdrawals attributable to a lack of efficacy and lower incidence of asthma exacerbations were observed with all active treatment groups. Latest guidelines2 3 define asthma severity and control in terms of future risk of adverse events, and acute exacerbations, as well as current symptoms and lung function. However, it should be noted that the relatively short treatment duration used in our study (8 weeks) may preclude conclusions regarding the potential benefit of FF in reducing the future risk of acute exacerbations. Studies of longer duration are required to evaluate the longer-term effects of FF on exacerbation frequency. An additional limitation of our study is that although we demonstrated efficacy with FF relative to placebo and acceptable tolerability and safety at doses <800 μg, the strict eligibility criteria used in our study excluded patients with severe asthma, thus restricting our generalisations to patients with moderate persistent asthma.
Once-daily FF was well tolerated during this study with a low overall incidence of on-treatment adverse events and generally similar to twice-daily FP. The incidence of oral or oropharyngeal candidiasis and urinary cortisol suppression was however higher in the 800 μg FF group compared with the lower FF dose groups. Pharmacokinetic and urinary cortisol analyses indicate this may have been attributable to higher systemic penetration of FF at the highest dose.
The results of this 8-week study in patients with moderate persistent asthma confirm the 24 h efficacy and tolerability of once-daily evening dosing of inhaled FF and indicate that once-daily doses of FF <800 μg have a favourable therapeutic index in this population. The absence of an efficacy dose–response suggests that FF 200 μg is suitable for further development as a monotherapy for the treatment of persistent moderate asthma or in a once-daily fixed-dose combination with the LABA vilanterol trifenatate.
References
Supplementary materials
Web Only Data thoraxjnl-2011-200308
Files in this Data Supplement:
Footnotes
Funding This study was funded by GlaxoSmithKline (study number FFA109684). Editorial support in the form of development of the draft outline, development of the manuscript first draft, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing and graphic services was provided by Geoff Weller at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline. The colour print publication fee was paid by GlaxoSmithKline.
Competing interests WWB has served as a consultant to AstraZeneca, Boehringer Ingelheim, Novartis and TEVA; served on advisory boards for Altair, Amgen, Centocor, GlaxoSmithKline, Johnson & Johnson, Merck Sharpe and Dohme and Pfizer; received lecture fees from Merck Sharpe and Dohme; and received research funding from AstraZeneca, Ception, GlaxoSmithKline, MedImmune and Novartis. ERB has served as a consultant to GlaxoSmithKline; and has performed clinical trials for GlaxoSmithKline, which have been administered by his employer Wake Forest University Health Sciences. EDB has served as a consultant to and received lecture fees from GlaxoSmithKline; and his institution has received remuneration for participation in clinical trials sponsored by GlaxoSmithKline. JL has served as a consultant to and received lecture fees from AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis and UCB Pharma; has been partly covered by some of these companies to attend previous scientific meetings including the ERS and the AAAAI; and has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, and Novartis. RF, AMD, LJ and BH are employees of and hold stock in GlaxoSmithKline. AW has served as consultant to Almirall, AstraZeneca, Chiesi, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis and Schering Plough; and has received research grants and travel expenses for attendance at ATS and ERS meetings from GlaxoSmithKline.
Ethics approval IRB ethics committee for each centre.
Provenance and peer review Not commissioned; externally peer reviewed.