Article Text

Abstract
Background Previous studies have shown that the incidence of idiopathic pulmonary fibrosis (IPF) is rising in the UK and USA. Death registrations and primary care data were used to determine the current trends in IPF incidence in the UK. Because routine clinical data sets were used, the term IPF clinical syndrome (IPF-CS) is used to describe individuals in this study.
Methods Age- and stratum-specific death registration rates between 1968 and 2008 were calculated and these were applied to the 2008 population to generate annual standardised expected number of deaths. Annual mortality rate ratios were calculated using Poisson regression. Computerised primary care records were used to determine incidence rates of IPF-CS between 2000 and 2008 stratified by age, sex and geographical region, and survival rates between calendar periods were compared.
Results Annual death certificate recording of IPF-CS rose sixfold across the study period from 0.92 per 100 000 in the 1968–1972 calendar periods to 5.10 per 100 000 in the 2006–2008 calendar period, and were higher in men and the older age groups. The incidence of IPF-CS in primary care increased by 35% from 2000 to 2008, with an overall incidence rate of 7.44 per 100 000 person-years (95% CI 7.12 to 7.77). Incidence was higher in men, the older population and in Northwest England.
Conclusions The incidence of IPF-CS in primary care and registered deaths from this cause in the UK continues to rise in the 21st century. The current findings suggest that there are >5000 new cases diagnosed each year in the UK.
- Interstitial lung disease
- clinical epidemiology
- pulmonary fibrosis
- interstitial fibrosis
Statistics from Altmetric.com
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common of the idiopathic interstitial pneumonias. Previous studies have reported that the mortality and incidence of IPF in the UK is on the rise.1–3 Current estimates of the incidence of IPF in the UK and the USA are 4.6 per 100 000 person-years and 6.8 per 100 000 person-years, respectively, suggesting that the disease is more common than originally thought.3 4 In contrast to cancer diagnoses, there is no mandatory registration of IPF diagnoses in the UK or elsewhere in the world. This issue is further compounded by the fact that the boundary between the disease entities within pulmonary fibrosis is not always clear clinically,5 making diagnoses challenging and raising speculation of whether they are temporal manifestations of the same condition.6 For this reason, epidemiological studies of population trends for IPF need to make the best use of routinely available clinical data sets to undertake research, to give pragmatic, real-world estimates of disease incidence, guide health service planning and provide insights into aetiology.
For the purpose of this epidemiological study we have chosen to use the term IPF clinical syndrome (IPF-CS) to describe the individuals included in our study. The aims of the study are to estimate mortality and incidence rates of IPF-CS stratified by age, sex and geographical region in the UK and investigate if prognosis had improved over time.
Methods
In this study, we used two different data sets to investigate whether the incidence of pulmonary fibrosis in the 21st century is still increasing in the UK. The first is routine death certificate data, and the second, a computerised longitudinal general practice database. Although the validity of pulmonary fibrosis diagnoses has previously been proven in both these data sets,1 7 8 it is likely that they will encompass a spectrum of individuals with fibrotic lung disease of unknown origin, and may also contain individuals with fibrotic non-specific interstitial pneumonia (NSIP) as well as IPF. The diagnostic classifications used in both data sets do not include these subclassifications and it is possible that these diseases have been labelled as IPF.
Data collection
The first data set was routine mortality data from the Office for National Statistics (ONS), derived from registered death certificates and coded for the underlying cause of death. This data set is available on CD-ROM and can be obtained electronically from the website of the ONS,9 and covers deaths that occurred in England and Wales from 1900 onwards. Our second data set was extracted from The Health Improvement Network (THIN) which is a computerised longitudinal primary care database recorded by UK general practitioners (GPs) as part of routine clinical care. Information is recorded from face to face consultations and following communication from secondary hospital referrals. THIN has been regularly used for the estimation of incidence and mortality from chronic diseases.10
Death registration analysis
Annual numbers of deaths for IPF-CS were obtained for the years 1968–2008 stratified by age and sex. Over this period of time, three different ICD (International Classification of Diseases) codes were used to code death from IPF-CS. From 1965 to 1978, ICD-8 code 517 (other chronic interstitial pneumonia), from 1979 to 1999, ICD-9 codes 516.3 (idiopathic fibrosing alveolitis) and 515 (postinflammatory fibrosis), and from 2000 onwards, ICD-10 code J84.1 (idiopathic pulmonary fibrosis). We used the total population from England and Wales for each year for the years 1968–2008 as our denominators.
For these analyses, the data on registered deaths and the general population were grouped into 5 year age bands over the age of 55 years and calendar periods were grouped into ∼5 year periods, but without combining years with different ICD codes. We calculated age- and sex stratum-specific mortality rates for each year and then applied these rates to the 2008 population to generate annual standardised mortality numbers. We also performed a subanalysis of the two different ICD-9 codes for IPF-CS (ICD-9 code 516.3 and ICD-9 code 515), where we calculated age- and sex-specific mortality rates for the years 1979–2000.
Poisson regression modelling was used to estimate annual mortality rate ratios with adjustment for sex and age. A multiplicative interaction term was used to test for interactions between annual trends in mortality rate ratios of IPF-CS and age or sex.
Incidence analysis
The version of THIN used in this study includes all information available up to July 2009 from 446 general practices. The Read codes H563.00 (idiopathic fibrosing alveolitis), H563.11 (Hamman–Rich syndrome), H563.12 (cryptogenic fibrosing alveolitis), H563100 (diffuse pulmonary fibrosis), H563z00 (idiopathic fibrosing alveolitis NOS) were used to identify individuals with a new diagnosis of IPF-CS made after 1 January 2000. The mid-year populations for each year were used as our denominators.
People were included in the cohort if they had at least one recorded IPF-CS diagnosis, their first diagnosis was recorded at least 12 months after their start date and they were aged at least 40 years when they received the first diagnosis. This definition was used to capture incident cases of IPF-CS and the age cut-off was used to improve diagnostic specificity. We excluded individuals with a co-existing diagnosis of connective tissue disease (see online appendix 1). We also excluded those with a co-existing diagnosis of extrinsic allergic alveolitis, sarcoidosis, pneumoconiosis and asbestosis (see online appendix 2), because it is not clear in this subset which diagnosis was correct, and this approach provided a more conservative estimate of disease incidence.
We calculated crude annual incidence rates, stratified by sex and age in 5 year age bands over the age of 55 years. Poisson regression modelling was applied to estimate annual incidence rate ratios, controlling for age, sex and health authority. Multiplicative interaction terms were again used to test for effect modification by age and sex. Due to the smaller size in this data set, we recoded the age groups into individuals under the age of 70 years and those aged ≥70 years when testing for interactions.
For our survival analysis, individuals with IPF-CS were assigned a start date that was defined as the date of diagnosis and a stop date that was defined as the earliest of either the last THIN data collection or the date of death. In order to determine if mortality was changing over time, we calculated the median survival for this cohort overall and survival stratified by 2 year calendar periods using Kaplan–Meir methods. Cox regression was used to compare mortality rates between the different age groups, sex and calendar periods. Multiplicative interaction terms were applied to test for interactions between mortality rates in the different calendar periods and age or sex.
Stata version 11 was used for all statistical analyses and likelihood ratio tests for all the hypothesis tests.
Results
Death certificate registration analysis: ONS data
There were 56 675 deaths attributed to IPF-CS in England and Wales from 1968 to 2008. The number of recorded deaths increased from 479 (659 deaths after standardisation to the 2008 population) in 1968 to 3019 in 2008 (see figure 1). The overall mortality rate standardised to the 2008 UK population over this period of time was 2.54 per 100 000 person-years (95% CI 2.52 to 2.56), with the rate increasing from 0.92 (95% CI 0.87 to 0.96) per 100 000 person-years in the 1968–1972 calendar period to 5.10 (95% CI 5.00 to 5.19) per 100 000 person-years in the 2005–2008 calendar period. Mortality rates were higher in men and the older population (see table 1). After controlling for the effects of sex and age, we calculated that the overall year on year increase in mortality was ∼5% (rate ratio 1.05, 95% CI 1.04 to 1.05). This equates to a sixfold increase in mortality over the calendar period studied.

Estimated number of deaths from idiopathic pulmonary fibrosis clinical syndrome, age standardised to the 2008 population of England and Wales. ICD, International Classification of Diseases.
Standardised mortality rates and Poisson regression modelling of deaths from idiopathic pulmonary fibrosis clinical syndrome (using the ONS data set)
There was evidence of a statistical interaction to suggest that the increase in mortality rates over time was higher in the older population (p<0.0001) and in men (p<0.0001). The age group-specific mortality rate ratios were 1.001 (95% CI 0.999 to 1.005) for individuals under the age of 55 years, 1.015 (95% CI 1.011 to 1.019) in individuals between 55 and 59 years, 1.024 (95% CI 1.021 to 1.026) in individuals aged 60–64 years, 1.043 (95% CI 1.041 to 1.045) in individuals between 70 and 74 years, 1.055 (95% CI 1.053 to 1.057) in individuals between 75 and 79 years, 1.072 (95% CI 1.070 to 1.074) in individuals aged 80–85 years and 1.098 (95% CI 1.095 to 1.10) in individuals who are ≥85 years. Despite having a statistically significant interaction, the mortality rate ratios in men and women were similar, with rate ratios of 1.050 (95% CI 1.049 to 1.051) and 1.049 (95% CI 1.048 to 1.051), respectively.
Subanalysis of the ICD-9 codes: ONS data
For the years 1979–2000, where two ICD-9 codes were used to indicate a death from IPF-CS, there were 12 989 deaths coded as being due to postinflammatory fibrosis (ICD-9 code 515) and 16 989 deaths coded as being due to idiopathic fibrosing alveolitis (ICD-9 code 516.3). Crude mortality rates for both ICD-9 codes increased with time, and were again higher in men and in the older age groups (see table 2).
Subanalysis of the ICD-9 codes for idiopathic pulmonary fibrosis clinical syndrome (using the ONS data set)
After controlling for age and sex, the estimated annual increase in mortality was marginally higher in idiopathic fibrosing alveolitis (rate ratio 1.05, 95% CI 1.05 to 1.06) compared with postinflammatory fibrosis (rate ratio 1.03, 95% CI 1.03 to 1.04). In these subgroups, there was no evidence of a statistical interaction to suggest that the increasing mortality trends were higher in the older groups or in men.
Incidence analysis: THIN data
We excluded 318 individuals with co-existing connective tissue disease, who had rheumatoid arthritis (211), systemic sclerosis (39) or other connective tissue diseases (68). We also excluded 134 individuals who had co-existing diagnoses of extrinsic allergic alveolitis (35), asbestosis (56), pneumoconiosis (14) and sarcoidosis (29). This left a total number of incident cases across the study period of 2074, which equates to a crude overall incidence rate of 7.44 per 100 000 person-years (95% CI 7.12 to 7.77). The majority of new cases (63%) were in men. The mean age at time of diagnosis was 74.3 years (SD 64.2–84.4). Incidence rates were found to be higher in men compared with women, the older population and in the North and West regions of the UK (see table 3).
Incidence rates and Poisson regression modelling of idiopathic pulmonary fibrosis clinical syndrome incidence (using the THIN data set)
After adjusting for age, sex and health authority, the estimated annual increase in the incidence of IPF-CS was 5% (rate ratio 1.05, 95% CI 1.03 to 1.06). There was no evidence of a statistical interaction to suggest that the annual increase in incidence over time was higher in men (p=0.07) but there was evidence of a statistical interaction to suggest it was higher in older age groups (p=0.011) The age group-specific annual incidence rate ratios were 1.01 (95% CI 0.99 to 1.04) in individuals under the age of 70 years and 1.08 (95% CI 1.06 to 1.11) in individuals aged ≥70 years.
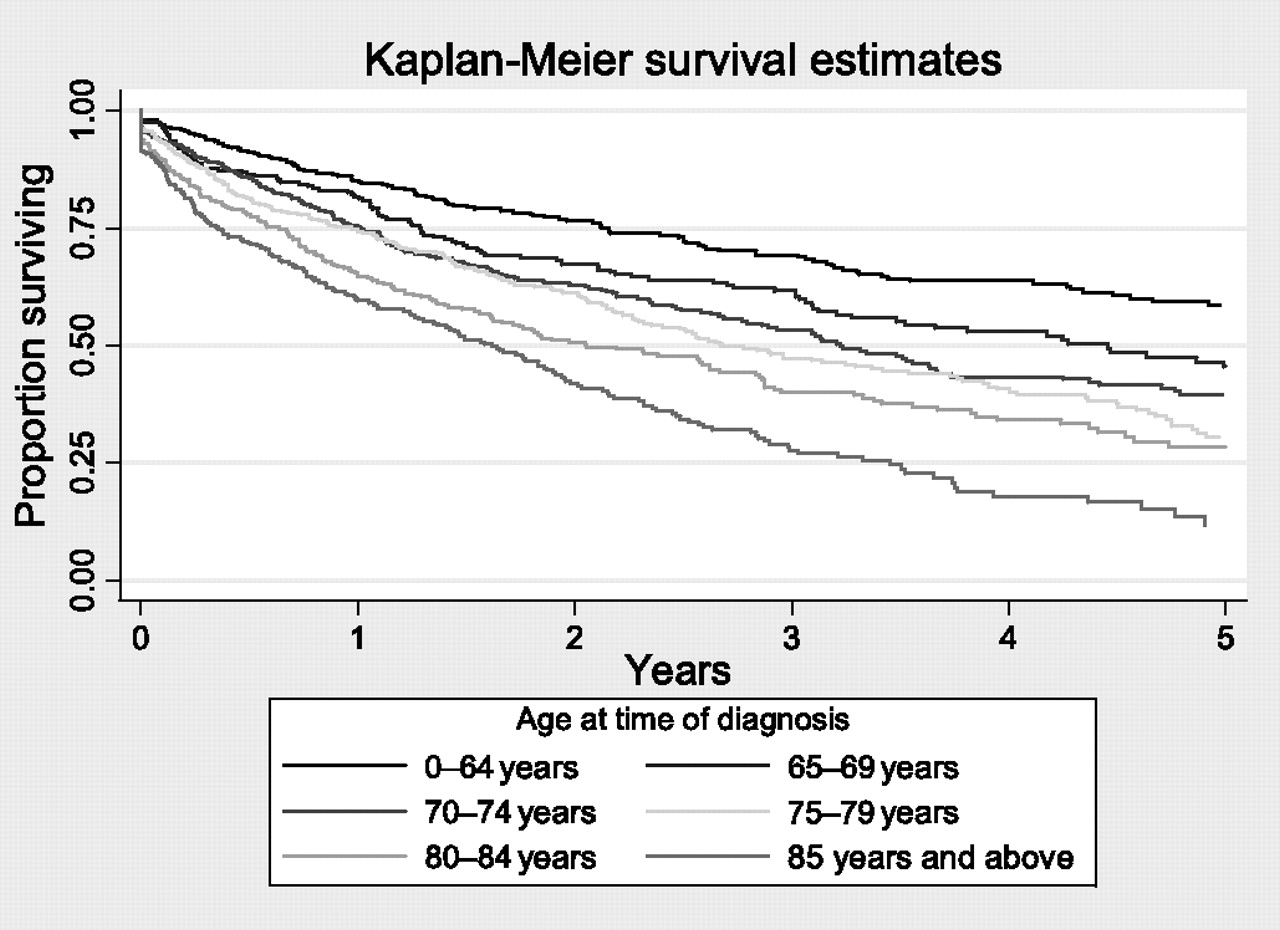
The mean follow-up period after diagnosis for our cohort was 2.2 person-years and, during this period, 1060 individuals (51%) died. The crude mortality rate for people with IPF-CS in this cohort was 228.8 per 1000 person-years (95% CI 193.8 to 216.7). Overall median survival for our cohort was 3.03 years. The median survival increased marginally from 2.6 years in 2000–2001 to 3.1 years in 2006–2008 (see table 4). This difference in survival between calendar periods was not statistically significant (p=0.10). We estimated the 5-year survival percentage of our cohort to be 37%. Within our cohort, male sex and increasing age were markers of poor prognosis (see table 4, figure 2). Median survival declined with increasing age. The estimated median survival of individuals under the age of 65 was 6.8 years. In individuals aged 65–69 years, 70–74 years, 75–79 years, 80–84 years and ≥85 years, median survival was 4.5, 3.2, 2.6, 2.0 and 1.6 years, respectively. The differences in survival between age groups was statistically significant (p<0.0001).
Survival analysis for individuals with idiopathic pulmonary fibrosis clinical syndrome (using the THIN data set)
{kind=link}
{kind=link}
Kaplan–Meier survival estimate stratified by age at time of diagnosis for the idiopathic pulmonary fibrosis clinical syndrome cohort (using the THIN (The Health Improvement Network) data set).
Discussion
Using two very different routinely available clinical data sets, we have found that the incidence of IPF-CS has increased over time. In both data sets, the size of the increase was 5% per year and this was not explained by ageing of the population. We found both death registrations and disease incidence to be higher in men and in older age groups, which is consistent with previous studies.1–3 Subanalysis of the ICD-9 codes also showed similar trends in both ICD-9 codes. The incidence of IPF-CS recorded in general practice was highest in Northwest England, Northern Ireland and Scotland. The prognosis of individuals in our cohort was poor and is consistent with previous studies.3 5 11–14 Overall median survival within our cohort improved slightly from 2.6 years in 2000–2001 to 3.1 years in 2006–2008. On the basis of our findings, we expect >5000 new cases of pulmonary fibrosis to be diagnosed annually in the UK.
The main strength of our death certificate registration analysis is the large numbers of registered deaths and the long period of time over which this information was obtained. This means that we have been able to give a precise estimate of mortality rates from the disease and to stratify our results by age, sex and calendar period. In addition, our incidence analyses used large computerised GP records, allowing us to give accurate estimates of incidence, stratified by age groups, sex, calendar periods and region. Studies of this nature and magnitude are not possible using only cases collected at centres in secondary care. The main potential weakness of our study, however, is the validity of the diagnosis of pulmonary fibrosis. Out of necessity, we have used available clinical data sets for our research and by their nature these do not include standardised diagnostic criteria for diagnosing pulmonary fibrosis. It seems unlikely to us, however, that a clinician will record a diagnosis of pulmonary fibrosis on either a death certificate or in a primary care record without some confirmation from secondary care. Support for this view comes from an earlier study which showed that the diagnostic accuracy of death certification of cryptogenic fibrosing alveolitis (CFA) was high.1 Of note, only 60% of patients with known CFA had this disease recorded on their death certificate1 which suggests that using death certification to estimate the incidence of IPF-CS will underestimate disease incidence by ∼40%. If we take this into account, we predict that the true mortality rate from IPF-CS between 2005 and 2008 would actually be ∼8.5 per 100 000 person-years, and this is very close to our estimate of disease incidence from general practice data for the same calendar period, which was 8.2 per 100 000 person-years. We have also previously shown that diagnoses of pulmonary fibrosis recorded in general practice data sets are accurate.8 More recently, Fell et al demonstrated that age is a strong predictor of IPF in patients with atypical radiographic appearances of usual interstitial pneumonia (UIP), whereby individuals over the age of 65 years had 89% specificity for confirming IPF by surgical lung biopsy.5 In both the data sets used in our study, >70% of the individuals were over the age of 65, reaffirming our belief that the majority of our cohort have UIP.
Another potential weakness is the possibility of coding errors when two ICD-9 codes were used. However, subanalysis of the individual ICD-9 codes showed similar trends, making it unlikely that the overall mortality trends over this period were due to diagnostic transfer or errors in disease coding. Despite the robust study design, there is a possibility that some of the cases in our GP cohort may have been prevalent rather than incident cases. This error is unlikely to have a major impact on incidence trends over time, but may have caused an overestimation of survival. However, our estimated median survival is not significantly longer than that of previous studies, which suggests that, if present, any misclassification is likely to be minor.11
There is limited information on age stratum-specific median lengths of survival, as many studies have lacked the statistical power to answer this question. However, where available, age-specific median survival time has been broadly similar to our findings,12–15 reiterating that survival is dependent on age.2 3 11–16 Araki and colleagues showed that median survival in individuals with biopsy-proven IPF under the age of 75 was ∼5.5 years.15 Similarly King et al demonstrated that survival in patients with biopsy-proven IPF under the age of 50 was significantly longer at 9.7 years.13 It is not surprising that younger individuals survive longer than older adults, but one possible explanation for why there is a difference in survival is the inclusion of individuals with NSIP and other chronic fibrotic lung diseases, which may carry a better prognosis than IPF, and might be more common within the younger age groups. However, the overall median survival of our cohort was ∼3 years, which is similar to other population studies of IPF,3 11 12 16 suggesting that diagnostic contamination, if present, was minimal.
Due to the difficulties in establishing accurate diagnoses in epidemiological studies, there are only a few large studies on the incidence of IPF. In the USA, a healthcare claims database and, more recently, a medical record linkage system have been used to estimate incidence rates,4 16 both of which showed similar rates to ours. Our study is consistent with our previous research in showing a progressive rise in the incidence of chronic fibrotic lung disease of unknown aetiology. Since IPF is the most common cause of this disease spectrum, it seems likely that a progressive increase in the incidence of IPF is central to this. We cannot be certain whether the observed increase in incidence represents a true increase, or is due to other explanations, or possibly both. The rapid nature of the increase suggests that an increase in disease ascertainment is also likely to be an important factor but, if this is the case, we would expect survival from this condition to improve over time, as milder cases are recognised as disease ascertainment moves towards 100%. This study provides no convincing evidence of this, suggesting that if increased ascertainment is an explanation, then levels of ascertainment are still far from complete. The introduction of high resolution CT in the early 1990s is important in the investigation and diagnosis of interstitial lung diseases, in particular IPF, but it did not appear to impact on the number of registered deaths.
The regional pattern in incidence which we observed is similar to those previously reported, with the highest incidence rates in North and West England as well as Scotland.3 It is not possible to say if these reflect true differences of aetiological importance or arise from variations in local primary or secondary care practice, and further work needs to be done in this area before any definite conclusions can be drawn.
In summary, IPF-CS is an important cause of respiratory mortality in the UK. Our results suggest that at present, 15 000 people in the UK have a diagnosis of IPF-CS, each year 5000 new cases are diagnosed and 5000 people with this disease spectrum will die. This means that in the UK, more people will die each year from IPF-CS than from ovarian cancer, lymphoma, leukaemia, mesothelioma or kidney cancer. At the moment we do not understand what causes IPF-CS, why the incidence is on the rise or how best to treat individuals with this disease spectrum. It is clear that IPF-CS is an important public health problem and warrants more research investment.
References
Supplementary materials
Web Only Data thx.2010.148031
Files in this Data Supplement:
Web Only Data thx.2010.148031
Files in this Data Supplement:
Footnotes
Funding VN is funded by a research grant from the Medical Research Council. RBH is funded by the GSK/BLF chair of Epidemiological Respiratory Research. JW and KMF are funded by an NIHR Clinician Scientist Fellowship.
Competing interests None.
Ethics approval This study was conducted with the approval of the Nottingham Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves