Article Text

Abstract
Background Intermittent viral exacerbations in patients with cystic fibrosis (CF) with chronic Pseudomonas aeruginosa (PA) infection are associated with increased bacterial load. A few clinical studies suggest that rhinoviruses (RV) are associated with the majority of viral-related exacerbations in CF and require prolonged intravenous antibiotic treatment. These observations imply that acute RV infection may increase lower respiratory symptoms by increasing planktonic bacterial load. However, the underlying mechanisms are not known.
Methods Primary CF airway epithelial cells differentiated into mucociliary phenotype were infected with mucoid PA (MPA) followed by RV and examined for bacterial density, biofilm mass, levels of chemokines and hydrogen peroxide (H2O2). The need for dual oxidase 2, a component of NADPH oxidase, in RV-induced generation of H2O2 in CF cells was assessed using gene-specific siRNA.
Results Superinfection with RV increased chemokine responses in CF mucociliary-differentiated airway epithelial cells with pre-existing MPA infection in the form of biofilm. This was associated with the presence of planktonic bacteria at both the apical and basolateral epithelial cell surfaces. Further, RV-induced generation of H2O2 via dual oxidase 2 in CF cells was sufficient for dispersal of planktonic bacteria from the biofilm. Inhibition of NADPH oxidase reduced bacterial transmigration across mucociliary-differentiated CF cells and the interleukin-8 response in MPA- and RV-infected cells.
Conclusion This study shows that acute infection with RV liberates planktonic bacteria from biofilm. Planktonic bacteria, which are more proinflammatory than their biofilm counterparts, stimulate increased chemokine responses in CF airway epithelial cells which, in turn, may contribute to the pathogenesis of CF exacerbations.
- Co-infection
- exacerbation
- oxidative stress
- biofilm
- airway epithelium
- bacterial infection
- cystic fibrosis
- respiratory infection
- viral infection
Statistics from Altmetric.com
- Co-infection
- exacerbation
- oxidative stress
- biofilm
- airway epithelium
- bacterial infection
- cystic fibrosis
- respiratory infection
- viral infection
Pulmonary manifestations due to chronic endobronchial infection are the leading cause of morbidity and mortality in individuals with cystic fibrosis (CF). Pseudomonas aeruginosa (PA), a principal pathogen in CF, chronically colonises the airways and often exists in a biofilm matrix. Despite chronic infection, patients with CF experience acute exacerbations only periodically, indicating that biofilm bacteria may act as a resilient reservoir for planktonic bacteria rather than a trigger for exacerbations. Consistent with this hypothesis, increased PA density was observed during episodes of acute exacerbations and required antibiotic treatment.1 Furthermore, PA isolated from sputum collected during exacerbations was similar to colonising flora by genotypic analysis, suggesting a clonal expansion of bacteria from the biofilm reservoir.2
The significance of respiratory viral infections in the pathogenesis of asthma and chronic obstructive pulmonary disease (COPD) exacerbations has long been recognised. Respiratory viral infection is implicated in 44–80% of asthma exacerbations and 46–50% of COPD exacerbations.3 Similarly, an improved method of viral detection suggested a significant role for viral infections in CF exacerbations.4–7 Respiratory viruses associated with CF exacerbations include influenza viruses A/B, rhinoviruses, respiratory syncytical viruses, parainfluenza viruses and adenoviruses.
Rhinoviruses (RV), which cause common cold, are also responsible for the majority of viral-related exacerbations in asthma and COPD.3 A few clinical studies have suggested that RV is also associated with the majority of viral-related exacerbations in CF.4 7–9 In one study, patients with RV infection required prolonged intravenous antibiotic treatment.10 In another study, RV infection was associated with increased use of antibiotics, prolonged hospitalisation and decline in lung function.8 Similarly, respiratory syncytial virus infection in patients with intermittent or chronic PA infection was associated with increased pseudomonal antibody levels.10 This circumstantial evidence suggests that viral infection may increase planktonic bacterial load which, in turn, may stimulate an intense inflammatory response and increase clinical symptoms that can be reduced by treatment with antibiotics. Consistent with this notion, in vitro studies suggested a more intense host inflammatory response to motile planktonic bacteria.11 12 However, the mechanisms by which viral infections increase the density of planktonic bacteria are not well known.
In the present study we examined whether superinfection with RV liberates planktonic bacteria and increases chemokine responses of CF airway epithelial cells preinfected with mucoid PA (MPA). We also investigated the mechanisms by which RV causes dispersal of planktonic bacteria from biofilm.
Methods
Full details are given in the online data supplement.
Rhinovirus
Rhinovirus serotype 39 was purchased from ATCC (Manassas, Virginia, USA). Viral stocks were generated and the tissue culture infectious dose (TCID50) was determined as described previously.13
Bacteria and growth conditions
A clinical isolate of MPA was grown in tryptic soy broth (BD Diagnostics, Sparks, Maryland, USA) and suspended in phosphate buffer solution (PBS).
CF cell cultures and infection
Primary CF airway epithelial cells were obtained from three patients with CF and grown either at the air–liquid interface to promote mucociliary differentiation or as monolayers.14 The use of CF bronchial segments was reviewed by the University of Michigan Institutional Review Board. Mucociliary-differentiated cells were sequentially infected with MPA (at multiplicity of infection of 0.01) followed by RV (1×106 TCID50), UV-irradiated RV or sham. Chemokine levels in the basolateral medium were determined by ELISA (R&D Systems, Minneapolis, Minnesota, USA). In some experiments the cells were preincubated with diphenylene iodonium (DPI) or 1140W (both from Sigma-Aldrich, St Louis, Missouri, USA), as indicated in the Results section. Cells treated with 0.1% dimethyl sulfoxide or media served as controls. Levels of lactose dehydrogenase (an index of cytotoxicity) were measured in the basolateral medium or cell culture supernatants from CF cell monolayers using Cytotox96 non-radioactive cytotoxicity kit (Promega, Madison, Wisconsin, USA).
Dispersal of biofilm
MPA biofilms were formed on the pegs of the MBEC assay system (Innovotech, Edmonton, Alberta, Canada) as described elsewhere.15 Briefly, the device consists of 96 conical pegs attached to a plastic lid and rests on a 96-well plate containing bacterial suspension (OD600 0.01) diluted in Lauria-Bertani broth and incubated for 48 h. The pegs with biofilm were rinsed with sterile PBS and exposed to conditioned media from CF airway epithelial cells for 30 min. The pegs were washed and stained with 0.2% crystal violet. Crystal violet bound to pegs was eluted with methanol and the optical density was measured at 590 nm to quantify the biofilm mass.
Measurement of hydrogen peroxide
Hydrogen peroxide (H2O2) produced from cells was measured using the Amplex red hydrogen peroxide/peroxidase kit (Invitrogen, Carlsbad, California, USA).
Transfection of cells
CF cells were transfected with dual oxidase 2 (Duox2) siRNA (Dharmacon, Lafayette, Colorado, USA) or non-targeting (NT) siRNA using Lipofectamine RNAiMAX (Invitrogen) by reverse transfection following the manufacturer's instructions. Reduction in expression of Duox2 was determined by real-time PCR in RV-infected cells using the forward primer 5′-AAC CTA AGC AGC TCA CAA CT-3′ and reverse primer 5′-CAG AGA GCA ATG ATG GTG AT-3′. The specificity of the product was determined by melting curve analysis.
Confocal microscopy
Cultures were fixed in methanol and incubated with a mixture of antibodies to PA (Abcam, Cambridge, Massachusetts, USA) and zona occludins (ZO)-1 (BD Biosciences, San Jose, California, USA).
Scanning electron microscopy
Cell cultures were processed for scanning electron microscopy as described previously.14
Statistical analysis
Results are expressed as mean±SEM or median (range). To compare groups, one-way analysis of variance (ANOVA) with Tukey–Kramer post hoc analysis or ANOVA based on ranks with Dunn's post hoc analysis was performed, as appropriate.
Results
Superinfection with RV increases chemokine responses in MPA-infected CF airway epithelial cells
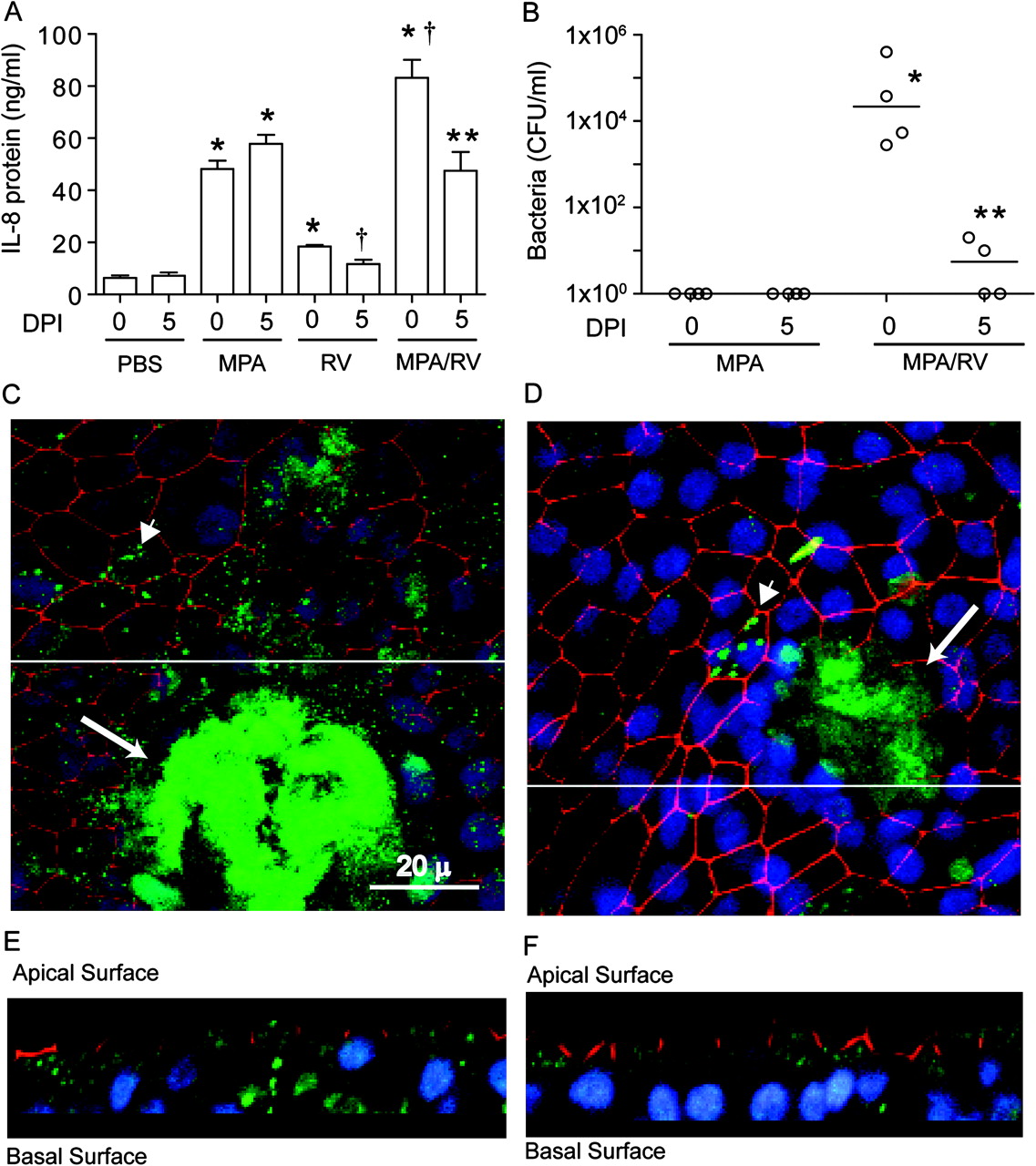
CF airway epithelial cells differentiated into mucociliary phenotype were infected with MPA or treated with PBS; 24 h later the cells were infected with sham, RV or replication-deficient UV-irradiated RV (UV-RV). Compared with cell cultures treated with PBS, cultures infected with MPA, RV or UV-RV showed significant increases in the chemokines interleukin 8 (IL-8), growth-regulated oncogene α (GRO-α) and epithelial cell-derived neutrophil-activating peptide 78 (ENA-78) (figure 1A–C). Cell cultures co-infected with MPA/RV showed synergistic increases in the levels of all three chemokines measured compared with cells infected with MPA or RV alone. This synergistic increase in chemokines was not observed in cells infected with MPA/UV-RV, indicating that an infectious virus is required for the observed increases.

Rhinovirus (RV) synergistically increases chemokine expression in cells infected with mucoid Pseudomonas aeruginosa (MPA). Mucociliary-differentiated cystic fibrosis (CF) airway epithelial cells were infected apically with MPA or treated with phosphate buffer solution (PBS) for 24 h. The cells were then infected with RV and incubated for another 24 h. Sham or ultraviolet-irradiated RV-infected cells (UV-RV) were used as controls. The following chemokine levels were measured in the basolateral medium: (A) interleukin 8 (IL-8); (B) growth-regulated oncogene α (GRO-α); (C) epithelial cell-derived neutrophil-activating peptide 78 (ENA-78). Data represent median and range calculated from two independent experiments performed in duplicate on cells obtained from three CF donors. Numbers within each panel represent medians calculated from six replicates for MPA-, RV- or MPA/RV-infected cells to show the synergistic increases. *p≤0.05 vs PBS-treated group; †p≤0.05 vs all other treatment groups (ANOVA on ranks with Dunn's post hoc analysis).
Small increases in both bacterial and viral loads were observed in cells co-infected with MPA and RV compared with cells infected with MPA alone or MPA/UV-RV (see tables 1 and 2 in online supplement). Together, these results suggest that the observed synergistic or additive chemokine responses depend on other factors besides small increases in bacterial and/or viral load.
RV infection facilitates transmigration of PA across mucociliary-differentiated CF airway epithelial cells
Cells infected with MPA/UV-RV or MPA/RV were immunostained with antibody to PA and ZO-1, a component of tight junction, and subjected to confocal indirect immunofluorescence microscopy. Basolateral media from these cultures were plated to determine the number of translocated bacteria. Confocal microscopy showed normal distribution of ZO-1 in the periphery of the cells and the presence of bacterial microcolonies (appearing as a green haze) with very few individual bacteria on the apical surface of MPA/UV-RV-infected cells (figure 2A). In contrast, MPA/RV-infected cell cultures showed dissociation of ZO-1 from the periphery of the cells in some areas and the presence of bacterial microcolonies along with numerous individual bacteria on the apical surface (figure 2B). Z-sections of these cultures revealed that, while bacteria were mainly on the apical surface in MPA/UV-RV-infected cultures (figure 2C), they were observed between the cells and at the basolateral surface in MPA/RV-infected cultures (figure 2D). Further, bacteria were recovered consistently (4.2×104±1.6×104 CFU/ml) in the basolateral media obtained from MPA/RV-infected cells, but not from MPA/UV-RV-infected cells. These results indicate that secondary infection with RV facilitates dispersal of bacteria from the microcolonies and transmigration of dispersed bacteria across the differentiated CF cultures, and this may potentially increase the interaction of bacteria with basolateral receptors.

Rhinovirus (RV) facilitates transmigration of mucoid Pseudomonas aeruginosa (MPA) across cystic fibrosis (CF) airway epithelial cells. Well-differentiated CF airway epithelial cells were sequentially infected with MPA followed by ultraviolet-irradiated RV (UV-RV) (A, C) or RV (B, D). Cells were immunostained with antibodies to a mixture of Pseudomonas aeruginosa (green) and zona occludin-1 (ZO-1; red) and counterstained with DAPI 4',6'-dimaino-2-phenylindole (blue). Images are representative of three independent experiments. (A, B) Apical view of cell cultures; (C, D) Z-section generated from the area marked with a white line in (A) and (B), respectively. Arrows in (A) and (B) represent bacterial microcolonies; arrowheads and asterisk in (B) represent two or three bacteria in a group and dissociation of ZO-1 from the periphery of cells, respectively.
RV infection causes dispersal of bacteria from MPA biofilm
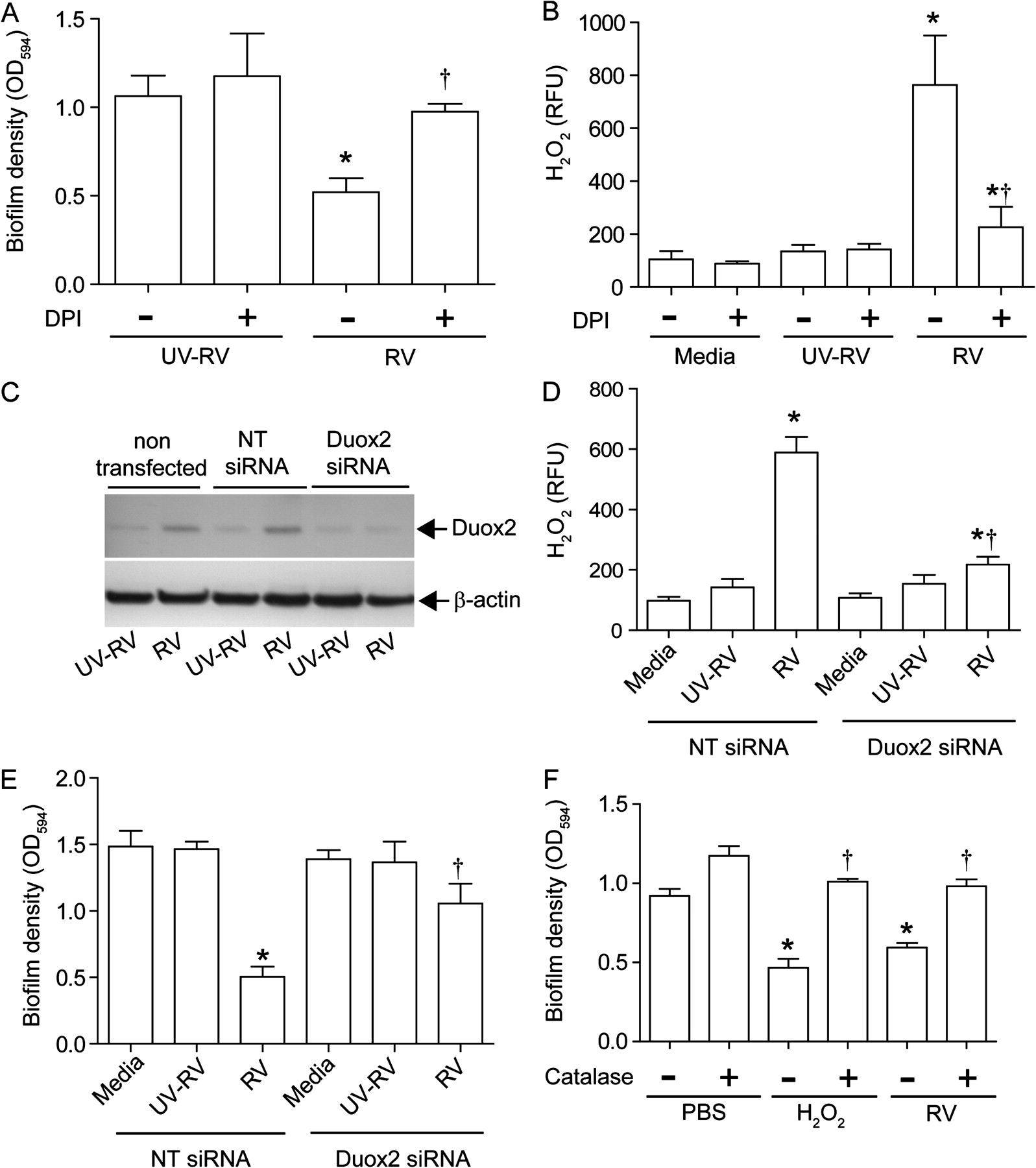
Scanning electron microscopy of MPA/UV-RV-infected mucociliary-differentiated cell cultures revealed a film covering the majority of the bacteria, suggesting the presence of bacteria in a biofilm (figure 3A). In contrast, MPA/RV-infected cultures showed hollows in the biofilm which were surrounded by bacteria that were not covered by a film, an indication of dispersal of the bacteria from the biofilm (figure 3B). These results imply that RV infection can disperse planktonic bacteria from biofilm.

Rhinovirus (RV) disperses bacteria from mucoid Pseudomonas aeruginosa (MPA) biofilm. Well-differentiated cystic fibrosis (CF) airway epithelial cells were sequentially infected with MPA followed by (A) ultraviolet-irradiated RV (UV-RV) or (B) RV and subjected to scanning electron microscopy. The asterisk and arrowhead represent MPA biofilm and individual bacteria; arrows in (B) point to hollows in the biofilm surrounded by bacteria not embedded in the biofilm, an indication of dispersed bacteria from biofilm. Images are representative of three independent experiments. (C) MPA biofilm grown on plastic pegs of the MBEC device were incubated with phosphate buffer solution, sham, RV inoculum or UV-RV and biofilm mass determined. (D) MPA biofilm grown on plastic pegs of the MBEC device were incubated with conditioned media from CF airway epithelial cells exposed to media, sham, RV or UV-RV and biofilm mass determined. Data represent mean±SEM calculated from four independent experiments performed in quadriplicate. *p≤0.05 vs conditioned media from cells exposed to media, sham and UV-RV (ANOVA with Tukey–Kramer post hoc analysis).
To determine whether RV is sufficient to disperse bacteria from biofilm, MPA biofilm grown on plastic pegs of an MBEC device (Innovotech) was exposed to PBS, cell culture media, RV inoculum equivalent to 1×106 TCID50 or UV-RV for 3 h and the biofilm mass left on the peg was quantified. None of these treatments affected the biofilm mass (figure 3C), suggesting that a factor(s) secreted by epithelial cells upon RV infection is responsible for dispersal of bacteria from biofilm. This led us to examine the effect of conditioned media from RV- or UV-RV- infected cells on biofilm density. In the subsequent experiments monolayers of CF primary cells were used because mucus present on the apical surface of differentiated cells interfered with the assay. Exposure of MPA biofilm grown on plastic pegs to conditioned media from RV-infected but not from UV-RV-infected cells decreased biofilm density (figure 3D). These results indicate that a factor(s) released from CF cells in response to infectious RV is responsible for biofilm disruption.
RV-induced H2O2 generation is sufficient for disruption of biofilm
RV has been shown to induce oxidative stress,16 17 and such environmental change can cause disruption of bacterial biofilm.18–20 We therefore examined the capacity of conditioned media from CF cells infected with RV or UV-RV in the presence of antioxidants to disrupt MPA biofilm. Treatment with DPI, an inhibitor of NADPH oxidase, completely reversed the capacity of conditioned media from RV-infected cells to disrupt biofilm (figure 4A and online figure 1A). In contrast, conditioned media from cells infected with RV in the presence of 1140W, an inhibitor of inducible nitric oxide synthase, had no effect on biofilm mass (see online figure 1C). Neither DPI nor 1140W alone had an effect on biofilm mass. Also, these inhibitors did not affect cell viability or viral load in the cells (see online figure 1E and 1F). These results indicate that DPI-inhibitable oxidative stress caused by RV is responsible for dispersion of biofilm bacteria.

Generation of hydrogen peroxide (H2O2) in cystic fibrosis (CF) airway epithelial cells in response to rhinovirus (RV) infection is responsible for bacterial dispersal from biofilm. (A) Conditioned media from monolayers of CF airway epithelial cells infected with ultraviolet-irradiated RV (UV-RV) or RV in the presence of 0 (−) or 5 μM (+) diphenylene iodonium (DPI) were incubated with mucoid Pseudomonas aeruginosa (MPA) biofilm and the biofilm mass was determined. (B) CF airway epithelial cells infected with UV-RV or RV or treated with media in the presence of DPI (0 or 5 μM) and H2O2 present in the media was measured. (C) CF cells were transfected with non-targeting (NT) or dual oxidase 2 (Duox2) siRNA, infected with RV or UV-RV and expression of Duox2 was determined by immunoblot analysis, representative of three independent experiments. (D) Levels of H2O2 in media from monolayers of CF cells transfected with siRNA specific to Duox2 or NT siRNA and infected with UV-RV or RV. (E) MPA biofilm grown on plastic pegs was exposed to conditioned media from cells transfected with Duox2 siRNA or NT siRNA followed by UV-RV or RV infection and biofilm mass was quantified. Cells treated with media served as control. (F) MPA biofilm was exposed to conditioned media from RV-infected cells or H2O2 (10 μM) in the presence or absence of catalase and biofilm mass was measured. Data represent mean±SEM calculated from four independent experiments performed in duplicates or quadriplicates. *p≤0.05 vs control; †p<0.05 vs RV-infected group in the absence of DPI or NT siRNA-transfected cells infected with RV (ANOVA with Tukey–Kramer post hoc analysis).
DPI inhibits flavoenzymes including NADPH oxidases. RV infection increased mRNA expression of Duox2, a component of NADPH oxidase, by 5.6±1.2-fold compared with sham or UV-RV-infected cells. Duox2 generates and secretes H2O2 directly into the extracellular milieu. In addition, RV infection also stimulated the generation of H2O2 in CF airway epithelial cells (12.6±5.81 μM), and this was inhibited by DPI (figure 4B and online figure 1B). To determine whether Duox2 stimulated by RV infection is required for generation of H2O2, monolayers of CF cells were transfected with non-targeting (NT) siRNA or siRNA specific to Duox2. CF cells transfected with siRNA specific to Duox2, but not NT siRNA, blocked the expression of Duox2 (figure 4C and online figure 2A and 2B) and inhibited H2O2 generation from RV-infected cells (figure 4D). In addition, conditioned media from Duox2 siRNA infected with RV was attenuated in its capacity to disrupt biofilm (figure 4E). This was not due to change in RV load in Duox2 siRNA transfected cells as there was no difference in RV load between NT-siRNA and Duox2 siRNA transfected cells (online figure 2C).
To further confirm that H2O2 induced by RV contributes to biofilm disruption, we initially determined the effect of H2O2 on the biofilm mass grown on plastic pegs. H2O2 decreased the biofilm mass in a concentration-dependent manner (online figure 3). Catalase, which neutralises H2O2, inhibited the reduction of biofilm mass caused by both H2O2 and conditioned media from RV-infected cells (figure 4F). These results confirmed that H2O2 generation induced by RV is required for disruption of biofilm.
DPI inhibits bacterial transmigration and IL-8 increase in differentiated CF cells superinfected with RV
To assess whether H2O2 induced by RV is required for the bacterial transmigration and observed synergistic increases in IL-8 production, well-differentiated CF airway epithelial cells were infected with PBS or MPA and incubated for 24 h. The cells were then infected with UV-RV or RV and incubated in the presence 0 or 5 μM DPI and the basolateral media were examined for IL-8 levels and the presence of bacteria. As observed earlier, in the absence of DPI, MPA/RV-infected cultures showed synergistic increases in IL-8 (figure 5A) and bacteria in the basolateral media (figure 5B). In contrast, MPA/RV-infected cells incubated in the presence of DPI showed a decreased IL-8 response, which was similar to the IL-8 response stimulated by MPA alone, and very few bacteria in the basolateral chamber. Confocal microscopy of cultures immunostained with antibodies to PA and ZO-1 showed the presence of biofilm along with numerous individual bacteria on the apical surface of MPA/RV-infected cell cultures in the absence of DPI (figure 5C). Z-sections of these cultures revealed the presence of bacteria on both the apical and basolateral surface (figure 5E). On the other hand, DPI-treated MPA/RV-infected cultures showed predominantly biofilm bacteria with very few individual bacteria on the apical surface (figure 5D), and Z-sections (figure 5F) showed bacteria on the basolateral surface only rarely. Together these results indicate that RV-induced DPI-inhibitable NADPH oxidase activity is responsible for dispersal of planktonic bacteria from biofilm, transmigration of bacteria and the related synergistic increase in IL-8 in CF airway epithelial cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diphenylene iodonium (DPI) inhibits bacterial transmigration and reduces interleukin 8 (IL-8) response in mucociliary-differentiated cultures sequentially infected with mucoid Pseudomonas aeruginosa (MPA) followed by rhinovirus (RV). Mucociliary-differentiated cystic fibrosis (CF) cultures were infected with MPA; 24 h later the cultures were infected with sham, ultraviolet-irradiated RV (UV-RV) or RV in the presence of DPI (0 or 5 μM) for another 24 h. (A) IL-8 concentration in the basolateral media determined by ELISA. (B) Number of bacteria in the basolateral chamber determined by plating. Data represent mean±SEM or median with range from three independent experiments performed in duplicate (*p≤0.05 vs media- or UV-RV-treated control groups; **p≤0.05 vs respective RV-infected group, ANOVA with Tukey–Kramer post hoc analysis or ANOVA on ranks with Dunn's post hoc analysis). †-p<0.05 vs all other groups. (C–F) Representative confocal images of cell cultures infected with MPA/RV in the presence of 0 or 5 μM DPI, respectively, showing bacteria (green), zona occludin-1 (red) and nuclei (blue). (C, D) Apical view of cell cultures. (E, F) Z-sections generated from an area marked with white lines in (E) and (F), respectively.
Discussion
In this study we show that superinfection with RV stimulates robust chemokine responses from CF airway epithelial cells preinfected with MPA. We also show that increased cytokine responses are associated with RV-facilitated dispersal of bacteria from MPA biofilm and transmigration of planktonic bacteria from the apical to the basolateral surface of mucociliary-differentiated CF airway epithelial cells. Furthermore, we provide evidence that RV infection induces H2O2 production in CF airway epithelial cells and this in turn causes dispersal of planktonic bacteria from biofilm. Together these results suggest that acute superinfection with RV increases chemokine responses and planktonic bacteria by causing dispersal of biofilm.
RV stimulates oxidative stress and chemokine responses from airway epithelial cells.16 21–23 It is therefore plausible that an RV-induced subtle change in the microenvironment of CF airways is sufficient to increase the pathogenesis of existing flora. Consistent with this notion, RV-induced mild oxidative stress appears to be sufficient for liberation of planktonic bacteria from the biofilm. Planktonic bacteria are motile and express virulence factors, and hence can stimulate inflammatory responses readily compared with relatively immotile biofilm bacteria.12 In CF airways, MPA persists in biofilm and is less stimulatory to airway mucosa due to ‘sandwich binding’.24 Dispersal of planktonic bacteria from the biofilm caused by RV infection may therefore lead to increased chemokine responses.
Following exposure to oxidative or other environmental stress, bacteria within the biofilm undergo coordinated dispersal events releasing free-swimming planktonic bacteria.18–20 RV, which can stimulate the expression of nitric oxide (NO) and H2O2,16 17 22 25 therefore can disperse planktonic bacteria from the biofilm. In the present study we found that RV-stimulated H2O2 mediated by Duox2, but not NO, is responsible for RV-induced liberation of planktonic bacteria from biofilm. Duox2 is one of the six homologues of NADPH oxidase catalytic phox subunit gp91phox 26 and bronchial epithelial cells express Duox2 in response to treatment with Th1 and Th2 cytokines, TLR3 agonist or infection with RV.27 Duox2 is located in the plasma membrane and can secrete H2O2 directly into the extracellular milieu,26 28 and thus can aid dispersal of bacteria from biofilm. Consistent with this notion, either inhibition of Duox2 expression or catalase, which neutralises the H2O2, abrogated the capacity of conditioned media from RV-infected cells to disrupt biofilm, suggesting that Duox2-mediated H2O2 generation plays a role in the dispersal of planktonic bacteria from the biofilm by RV. Lack of NO generation in CF cells in response to RV infection may be due to inducible nitric oxide synthase deficiency in these cells.29 30
In addition to dispersing bacteria from the biofilm, RV can also facilitate transmigration of bacteria across the airway epithelium because it has the capacity to compromise barrier function.31 32 This in turn may promote new interactions of dispersed planktonic bacteria with basolateral receptors which are otherwise not accessible, leading to increased production of cytokines from airway epithelial cells.
It is well established that bacteria in biofilms are more resistant to antibiotics than planktonic bacteria.33 In this regard, one can argue that disrupting biofilms to liberate planktonic bacteria that are more easily killed by antibiotics should be beneficial in CF where bacteria are thought to persist in biofilm. In addition, RV-induced H2O2 should also be beneficial in CF because H2O2 treatment has been proposed to kill planktonic bacteria.20 However, planktonic bacteria dispersed from biofilm are often as resistant to antibiotics as their biofilm counterparts34 and also express antioxidant enzymes such as alkyl peroxidase and catalase, each of which can neutralise H2O2 in the extracellular milieu.35 It is therefore conceivable that RV-mediated dispersal of planktonic bacteria from biofilm increases inflammation in CF, requiring prolonged hospitalisation and use of intravenous antibiotics.
In conclusion, our results suggest that RV-induced oxidative stress liberates planktonic bacteria from biofilm and facilitates transmigration of planktonic bacteria across the mucociliary-differentiated CF airway epithelial cells. This in turn may increase the interaction of planktonic bacteria with the basolateral receptors, leading to increased production of chemokines. These findings provide a novel mechanism by which RV infection may increase planktonic bacterial load and chemokine levels in CF.
Acknowledgments
The authors thank Sasha Meshinchi, Microscopy and Image Analysis Laboratory, University of Michigan, for his assistance with scanning electron microscopy.
References
Supplementary materials
Footnotes
Funding This work was supported by National Institutes of Health grants HL0897720 (USS), HL082550 and HL081420 (MBH) and the Cystic Fibrosis Foundation (USS).
Competing interests None.
Ethics approval Use of CF bronchial segments was reviewed by the University of Michigan Institutional Review Board (IRB number HUM00000230).
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves