Article Text

Abstract
Background In idiopathic pulmonary fibrosis (IPF) the distribution and spatial-temporal progression of fibrotic changes may be influenced by general or locoregional conditions. From this perspective, patients with asymmetrical disease (AIPF) may be unique.
Methods This retrospective study included 32 patients (26 men, mean±SD age 69±7 years) with AIPF, as defined by an asymmetry ratio (most affected – least affected fibrosis score)/(most affected + least affected fibrosis score) >0.2. The global fibrosis score was the average of the right and left scores. Patients with AIPF were compared with 64 matched controls with symmetrical IPF.
Results Patients with AIPF did not differ from controls in global fibrosis score and forced vital capacity, but carbon monoxide transfer factor was less decreased (52±19% vs 43±13%, p=0.009). The rate of gastro-oesophageal reflux and acute exacerbations was significantly higher in patients with AIPF (62.5% vs 31.3%, p=0.006 and 46.9% vs 17.2%, p=0.004, respectively). In patients with AIPF the right side was more likely to be involved (62.5%); the median asymmetry ratio was 0.5 (range 0.24–1). Although the global fibrosis score worsened significantly in all 23 patients with AIPF with serial high-resolution CT scans (p<0.0001), pulmonary fibrosis remained asymmetrical in all except three. During follow-up, 15 patients with AIPF experienced 18 acute exacerbations. The first episode was virtually unilateral, occurring in the most affected lung in 10 patients (66.7%). Survival was similar between patients with AIPF and controls.
Conclusion AIPF may be related to locoregional factors including gastro-oesophageal reflux which may be responsible for both disease expansion and the occurrence of acute exacerbations.
- Idiopathic pulmonary fibrosis
- asymmetry
- natural history
- acute exacerbation
- gastro-oesophageal reflux
- interstitial fibrosis
Statistics from Altmetric.com
- Idiopathic pulmonary fibrosis
- asymmetry
- natural history
- acute exacerbation
- gastro-oesophageal reflux
- interstitial fibrosis
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial pneumonia of unknown cause limited to the lungs and associated with the histological appearance of usual interstitial pneumonia (UIP). In the absence of surgical lung biopsy, a typical high-resolution CT (HRCT) scan is required for a definite diagnosis of IPF. According to the ATS/ERS consensus classification of idiopathic interstitial pneumonias, a typical HRCT picture consists of ‘bilateral, predominantly basal and peripheral reticular pattern with minimal ground glass opacities’, but there is no mention of the symmetry of abnormalities.1 To date, the clinical course of IPF is unclear but there are shreds of evidence suggesting that it may be less of a gradual decline and more of a step-like process, with periods of relative steadiness punctuated by episodes of acute exacerbations (AE)—that is, acute respiratory decompensations with no identifiable cause.2 These events may result in the majority of deaths related to IPF.2
In IPF the distribution of fibrotic changes between the two lungs and their spatial-temporal progression are unknown. These may be influenced by various general or locoregional conditions which could be shared by a gradual decline and AE or be independent of each other. From this perspective, patients presenting with asymmetrical IPF (AIPF) are intriguing and offer a unique opportunity to understand better the pathogenesis and natural history of IPF. First, in this context, an aetiological factor may exert its profibrotic or antifibrotic effect with partiality for one of the lungs. Second, patients with AIPF may have a distinctive clinical/functional/radiological presentation or a particular disease course because one lung is relatively preserved.
We report a case–control study of patients with AIPF. The objectives of the study were to describe patients with AIPF in terms of (1) clinical characteristics with a focused interest on gastro-oesophageal reflux (GER), (2) radiological and functional features, (3) outcome, including HRCT progression and AE episodes and (4) mortality, and to compare them with patients with symmetrical IPF.
Methods
Patients
This retrospective study was conducted in two centres, Avicenne Hospital and Louis Pradel Hospital. It received institutional review board approval and the requirement for informed consent was waived. Clinicians of the two participating centres were asked to report their cases with an a priori AIPF. Patients' medical records were then reviewed by three of the authors (CT, HN and VC) and they were included if the following criteria were met: (1) diagnosis of IPF according to ATS/ERS recommendations1 and (2) asymmetrical involvement as defined by an asymmetry ratio of >0.2 on the HRCT scan.
Inclusion was the date of the first HRCT scan showing significant asymmetry (diagnosis of AIPF). GER was defined according to the French consensus: typical clinical symptoms and/or oesophagitis > grade 1 on oesophagoscopy and/or positive 24 h ambulatory pH monitoring.3 AE was defined according to Akira's criteria.4
Controls
Controls were patients with symmetrical IPF as defined by an asymmetry ratio of ≤0.2. For each control the first and the last available HRCT scans were reviewed to ensure that the ratio remained ≤0.2 during follow-up. Two controls were matched with one patient for the year of IPF diagnosis.
HRCT analysis
Serial scans were independently reviewed by two radiologists (PYB and MB). Lung fields were divided into five levels5 and the following features were examined: ground glass opacification, reticulations (including intralobular reticulations, microcystic and macrocystic honeycombing) and emphysema. The extent of each feature was quantified as the proportion of involved parenchyma surface. A measure was assigned for each of the five sections (between 0% and 100%, censored at 5%). If the difference between readers was <20%, the mean value of the two estimations was taken; otherwise, a consensus measure was reached. For each feature, right and left scores represented the average of the five measures obtained for each lung. The global score was calculated as the average of the right and left scores. The fibrosis score was the summation of ground glass and reticulation scores.
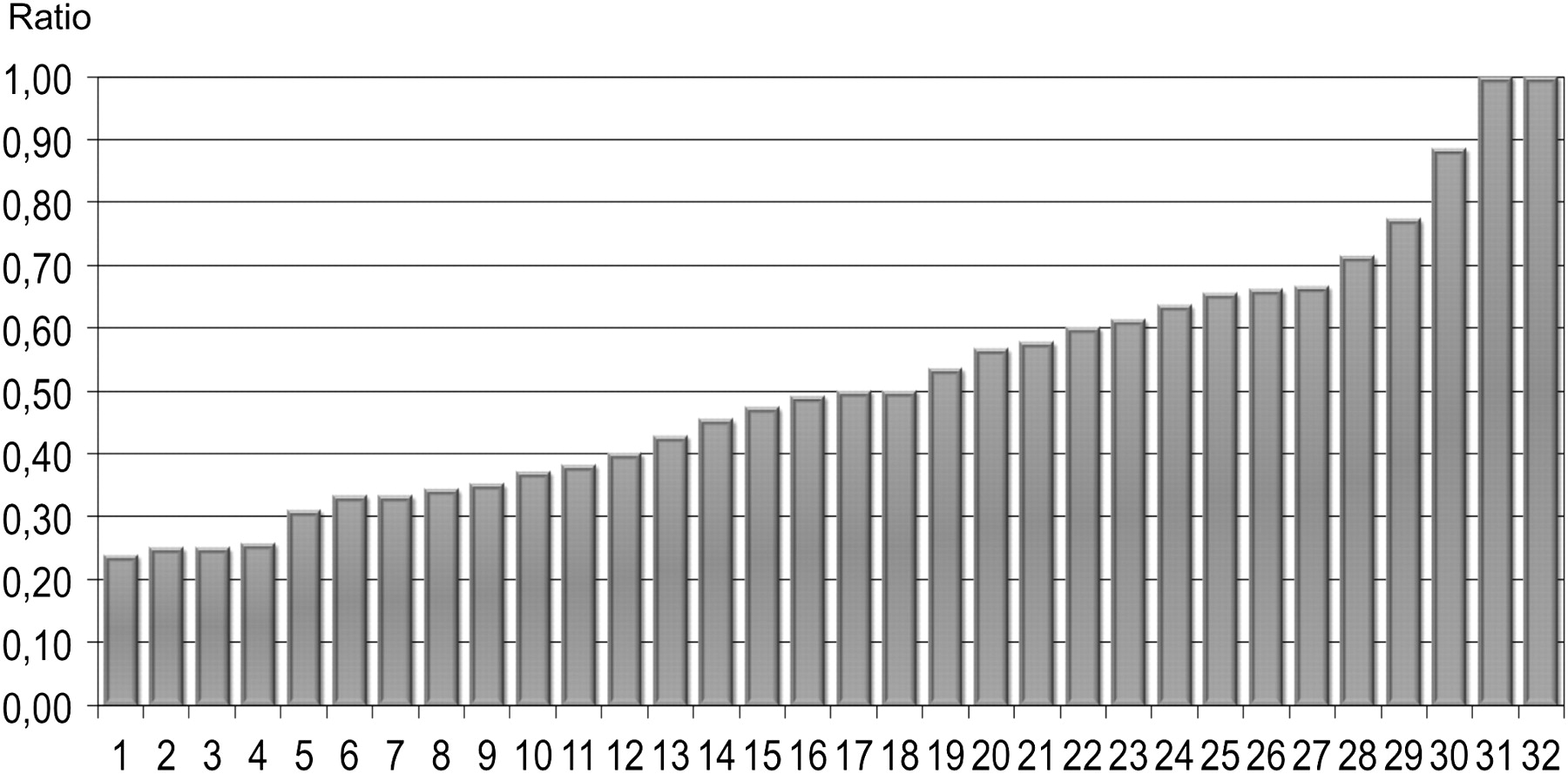
The asymmetry ratio was calculated as (most affected – least affected fibrosis score)/(most affected + least affected fibrosis score) and ranged from 0 to 1 (figure 1). The asymmetry ratio only reflects the imbalance of damaged surface between the two lungs, no matter the extent of fibrosis. A ratio of 0 corresponds to perfect symmetry between the two lungs and a value of 1 to strictly unilateral fibrosis; a value of 0.2, which was the arbitrary threshold chosen to define AIPF, means that the fibrosis score of the predominantly affected lung is 1.5 times greater than that of the other lung.

Evaluation of the asymmetry ratio. The extent of fibrosis (ground glass plus reticulation) was measured as the proportion of involved lung surface at each of five levels (between 0% and 100%, censored at 5%). Right and left fibrosis scores were the average of the five measures obtained for each lung. Asymmetry ratio was calculated as (most affected – least affected fibrosis score)/(most affected + least affected fibrosis score).
Statistical analysis
The results are expressed as percentages and mean±SD values. The reproducibility of imaging analysis was evaluated by the intraclass correlation coefficient on all pooled HRCT scans (ICC). The Pearson correlation coefficient was used to analyse the relationship between global fibrosis score and physiological parameters. The proportions of patients with a right predominance of pulmonary fibrosis and GER in the AE and non-AE groups were compared using χ2 tests. Forced vital capacity (FVC) and carbon monoxide transfer factor (Tlco) in the AE and non-AE groups were compared using unpaired t tests. A paired t test was used for comparing changes in HRCT scores during follow-up. Patients with AIPF were compared with controls by the χ2 test or the unpaired t test when appropriate. Survival of patients and controls was estimated by the Kaplan–Meier method (transplanted patients were censored at the date of transplantation) and compared with the log rank test. p Values <0.05 were considered significant.
Results
Thirty-nine cases recruited from January 1986 to July 2008 were reviewed; 32 patients were included (23 in Avicenne Hospital and 9 in Louis Pradel Hospital) and 7 were excluded (2 with no HRCT scan, 4 patients not fulfilling the ATS/ERS criteria for IPF and 1 with Sjogrën's syndrome). The 32 patients with AIPF were compared with 64 matched controls with symmetrical IPF.
Patient characteristics
The characteristics of the patients are summarised in table 1. The AIPF group consisted of 26 men and 6 women of mean±SD age 69±7 years at the time of AIPF diagnosis. Nine patients (28%) had an open lung biopsy. Pulmonary fibrosis was asymmetrical at presentation in 29 patients (90.6%) and was at first symmetrical in the remaining 3 cases (9.4%). At inclusion, pulmonary fibrosis was prominent in the right lung of 20 patients (62.5%) and in the left lung of 12 patients (37.5%). Information on GER symptoms was available in the medical records of all patients and oesophageal investigations were performed in 17 cases (oesophagoscopy, n=14; 24 h ambulatory pH monitoring, n=8; both oesophagoscopy and 24 h pH monitoring, n=5). GER was present in 20 patients (62.5%), as diagnosed by typical clinical symptoms (n=18/32) and/or oesophagitis on oesophagoscopy (n=7/14) and/or positive 24 h pH monitoring (n=5/8). Hiatal hernia was present in 11 cases and one patient suffered from a disabling nutcracker oesophagus. Nineteen patients were questioned about their sleeping habits: 3 had no preferred sleeping position and 15 of the 16 remaining patients (94%) used to fall asleep on the most involved lung side.
Patient characteristics at the time of diagnosis of AIPF and comparison with controls with symmetrical IPF
As indicated in table 1, patients with AIPF differed significantly from controls being older (69±7 vs 63±12 years, p=0.020) with a higher frequency of GER (62.5% vs 31.3%, p=0.006) and a less altered Tlco (52±19% vs 43±13%, p=0.009).
HRCT findings
Interobserver agreement
HRCT measures estimated by the two radiologists showed a good ICC (ICC 0.836–0.972 for reticulation scores and ICC 0.718–0.996 for ground glass scores depending on the analysed level), which were similar for the more and the less affected lungs.
HRCT findings at inclusion
HRCT findings are summarised in table 2. The distribution of patients with AIPF according to the asymmetry ratio is shown in figure 2. Median value of asymmetry ratio was 0.50, which corresponds to a fibrosis score of the predominantly affected lung three times higher than that of the other lung. Pulmonary fibrosis was strictly unilateral at inclusion in two cases. Emphysema was observed in nine patients (28%). The global emphysema score in these patients was 15.9±10.07% and ranged from 4% to 52%. Of note, four patients had a peculiar form of AIPF with prominent fibrosis in one lung and emphysema in the other; one patient had unilateral fibrosis and lone contralateral emphysema. A significant relation was found between the global fibrosis score and FVC% predicted (r=−0.52, p=0.009) but not between the global fibrosis score and Tlco% predicted (r=−0.25, p=0.319).
HRCT findings at the time of diagnosis of AIPF (n=32)

Distribution of patients with asymmetrical idiopathic pulmonary fibrosis according to the asymmetry ratio (n=32). The asymmetry ratio was 0.20–0.29 in 4 cases, 0.30–0.49 in 12 cases, 0.50−0.69 in 11 cases and 0.70−1.00 in 5 cases.
Comparisons between patients and controls confirmed a significantly higher asymmetry ratio in patients with AIPF (0.52±0.21 vs 0.05±0.06, p<0.0001). Conversely, the global fibrosis score was similar (25.7±11.9% vs 24.4±11.1%, p=0.600) as well as its two components: global reticulation score (18.6±9% vs 15.8±5.4%, p=0.060) and global ground glass score (6.5±7.3% vs 8.59±8.97%, p=0.230).
Evolution of HRCT during follow-up
Twenty-three patients with AIPF completed at least a second HRCT scan during follow-up, with the last available one being performed 32±26 months after inclusion. Figure 3 shows the evolution of the global fibrosis score and asymmetry ratio. The global fibrosis score worsened in all patients with a mean increase of 13.3±10.6% between the first and last HRCT scan (p<0.0001). Pulmonary fibrosis remained clearly asymmetrical in 20 patients (86.9%) and turned symmetrical in only three cases (13.1%) as a result of a marked bilateral AE. The asymmetry ratio was not significantly different between the first and last HRCT scans (p=0.830).

Evolution of global fibrosis score and asymmetry ratio on serial high-resolution CT (HRCT) scans of patients with asymmetrical idiopathic pulmonary fibrosis (n=23). Each patient may have undergone several HRCT scans during follow-up. (A) Evolution of individual values of global fibrosis score. The global fibrosis score worsened in all cases. (B) Evolution of mean±SD global fibrosis score from the first to the last available HRCT scan (22.7±8.9% vs 36.0±15.5%; p<0.0001, paired t test). (C) Evolution of mean±SD asymmetry ratio from the first to the last available HRCT scan (0.51±0.18 vs 0.50±0.18; p=0.830, paired t test).
Acute exacerbations
Fifteen patients with AIPF (46.9%) experienced an AE during follow-up (1 episode in 12 patients and 2 episodes in 3 patients, n=18 episodes). Patients with or without an AE were not significantly different with regard to the side of pulmonary fibrosis (right predominance: 8/15 vs 12/17, respectively, p=0.522), GER frequency (9/15 vs 11/17, respectively, p=0.927) and functional parameters (FVC: 62±19% vs 76±26%, p=0.108; Tlco: 53±17% vs 53±24%, p=0.984, for AE and non-AE groups, respectively). At the time of AE, the arterial oxygen tension was 53.7±14.9 mm Hg with a mean fall of 17.6±7.5 mm Hg compared with the previous measurement. HRCT scans were available for each episode of AE. AE was immediately bilateral in five patients (33.3%) who died rapidly. Ten patients (66.7%) originally presented with ground glass opacities, virtually unilateral and showing up in the most fibrotic lung. All survived this first AE but three of them suffered a fatal subsequent bilateral episode occurring 5, 6 and 24 months later (figure 4). The remaining seven patients were still alive 26.8±24 months after the AE.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
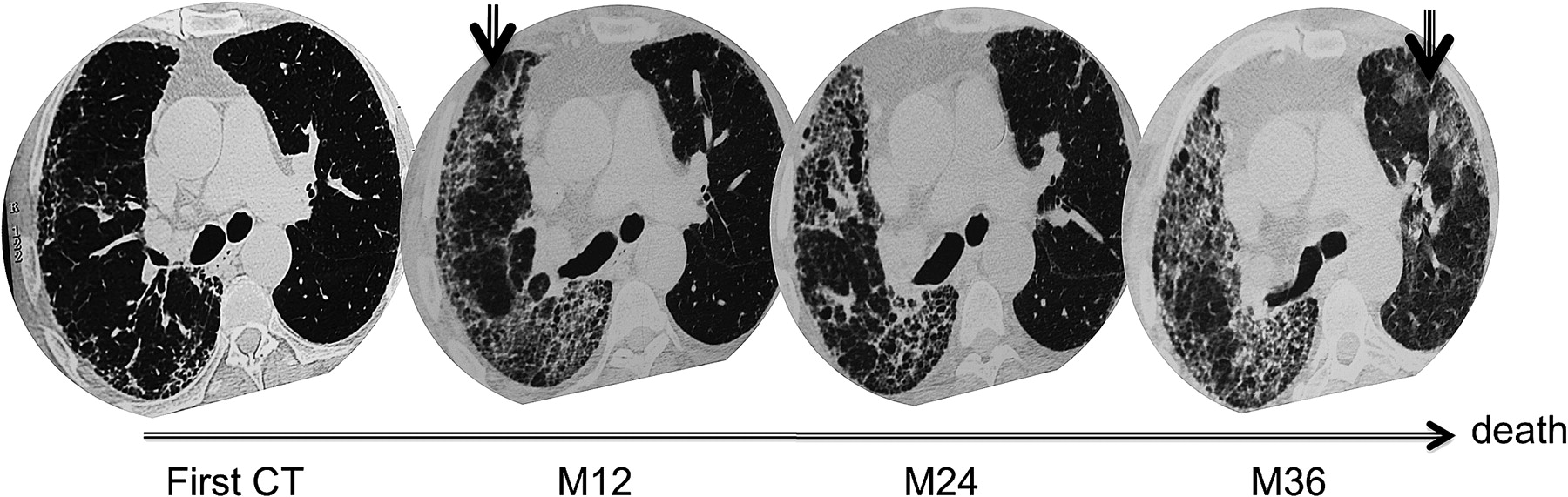
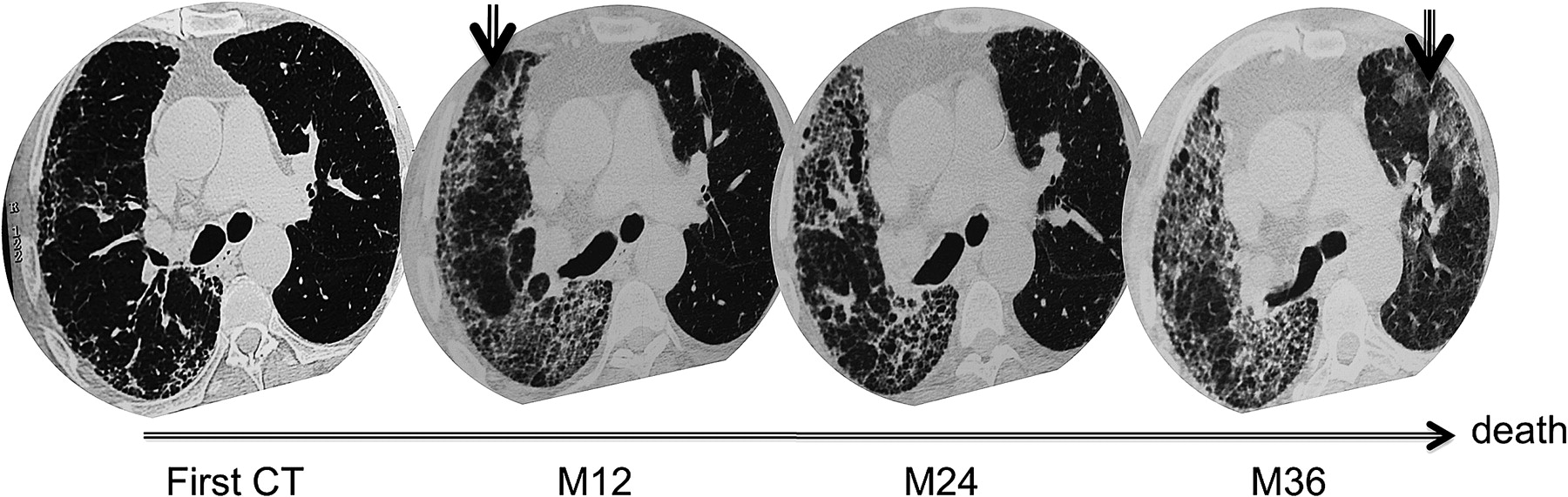
An example of two consecutive acute exacerbations (AE) in a patient with asymmetrical idiopathic pulmonary fibrosis. The baseline high-resolution CT (HRCT) scan shows that subpleural reticulations are predominant in the right lung. The HRCT scan taken 12 months later during the first AE shows superimposed unilateral ground glass opacities. After recovery there is a decrease in ground glass opacities, replaced in some areas by reticulations and honeycomb lesions. The HRCT scan during the second AE, which occurred 24 months after the first AE, shows bilateral ground glass opacities. The patient died of this second AE.
A follow-up HRCT scan was available after recovery for the 10 patients who initially presented with a unilateral AE and then showed replacement of ground glass opacities by reticulations (figure 4). The reticulation score after recovery/reticulation score during AE was significantly higher in the lung affected by AE than in the unaffected lung (1.88±0.12 vs 1.29±0.13, p=0.008).
AE was observed in 11 of the 64 controls (17.2%) during follow-up (1 episode in 9 patients and 2 episodes in 2 patients), which was strikingly lower than in the patients with AIPF (p=0.004). Of note, the AE was immediately bilateral in all cases and led to death in six patients (54.5%).
Mortality
Patients with AIPF were followed for 37±30 months and controls for 44±36 months (p=0.330). At the end of follow-up, 15 patients with AIPF were dead (46.9%), 1 was transplanted and 16 were alive. The causes of mortality were: AE (n=8), end-stage respiratory failure (n=3), pulmonary carcinoma (n=2), pneumothorax (n=1) and unknown (n=1). Survival of patients with AIPF was similar to that of controls, with a probability of survival of 75% vs 87%, 53% vs 63.3% and 50% vs 51.4% at 1, 3 and 5 years, respectively (p=0.331).
Discussion
We report here a series of patients fulfilling the diagnostic criteria for IPF but presenting with a frankly asymmetrical disease. Patients with AIPF significantly differed from those with symmetrical IPF with a much higher frequency of GER and AE. The asymmetry observed in a number of patients with IPF highlights the role of an aetiological factor such as GER or specific locoregional conditions in the development and/or progression of pulmonary fibrosis, including AE.
One may argue that asymmetrical lesions are related to fixed sequelae of a previous fibrotic condition. However, the fibrosis score worsened in all patients, supporting the idea of an ongoing fibrotic process. Moreover, pathological examination did not reveal end-stage fibrosis but a typical pattern of UIP in all patients who underwent an open lung biopsy. Finally, over time the pulmonary fibrosis remained frankly asymmetrical in the majority of patients, AE increased preferentially in the most affected lung and reticulations appeared in place of ground glass opacities. Taken together, these results suggest the existence of a common aetiological factor or locoregional conditions that are involved in both the progression of fibrotic disease and the occurrence of AE. This may be either ‘deleterious’ or ‘protective’ in an asymmetrical fashion.
Although controversial, a possible link between GER and IPF has long been considered.6–8 Patients with AIPF more frequently had overt GER than controls with symmetrical IPF (62.5% vs 31.3%, p=0.006). These frequencies are lower than expected since the prevalence of abnormal 24 h pH monitoring has been reported to reach 87% in a recent large prospective study of IPF.7 The true prevalence of GER may have been underestimated in both populations with symmetrical or asymmetrical disease. In fact, patients with GER do not necessarily present with typical gastrointestinal complaints6 and our patients or controls were not systematically investigated. In addition, some patients may suffer from alkaline reflux. Only prospective studies with new investigative tools such as pH impedance monitoring would provide an accurate answer on the role of GER, as recently published in scleroderma.9
However, in addition to the difference between patients with AIPF and controls, two other results point to the role of GER in AIPF. First, most of our patients with AIPF (62.5%) showed a predominance of fibrosis in the right lung, which is more likely to be directly damaged by repeated microaspirations. Second, the side of fibrosis, whether right or left, matched with the preferential posture to fall asleep in 94% of cases. Reflux events occur during both daytime and night-time hours but, it is well known that those occurring during sleep are significantly longer and predominate while patients fall asleep soon after the evening meal.10 11 Interestingly, the instillation of gastric juice into the right main bronchus of pigs resulted in alveolar injury followed by the development of asymmetrical pulmonary fibrosis.12 In this model, the causative agents were found to be both gastric hydrochloric acid and pepsin, whereas acidity was not necessary to initiate the fibrotic process.12 Furthermore, pepsin may promote epithelial mesenchymal transformation in vitro.13
Apart from GER, other conditions may explain the particular distribution and progression of pulmonary fibrosis seen in patients with AIPF. Cool et al have shown that the fibrolast foci seen in IPF have a highly complex reticulum extending from the pleura into the lung parenchyma.14 This concept is further supported by the study by Decologne et al in which transient transfer of the transforming growth factor β1 (TGF-β1) gene by adenoviral vectors to the pleural cavity of rats induced not only progressive pleural fibrosis but also pulmonary fibrosis.15 Pleural fibrosis, through a process involving mesothelial-fibromatoid transformation, expands into the lung parenchyma adjacent to the pleural surface from the visceral layer but not into the muscle from the parietal layer, suggesting that a distinct local environment is required for progressive fibrotic responses in the tissue.15 The connectiveness of fibrolast foci is closely intertwined with a vascular network and vascular perfusion/angiogenesis is well known to be an important mechanism of fibroproliferation and repair.16 In AIPF, the asymmetrical progression of pulmonary fibrosis may be due to previous pleural or vascular abnormalities but these were not comprehensively explored in our study. It is, however, noteworthy that a case of unilateral UIP has been published in a patient with ipsilateral slow-growing pulmonary artery sarcoma which resulted in chronic pulmonary ischaemia and systemic arterial supply.17 Similarly, another case has been reported with unilateral IPF in the right middle and lower lobe owing to congenital absence of the right interlobar pulmonary artery whereas the upper lobe was spared.18 Interestingly, an unusual form of AIPF was observed in four of our cases with prominent pulmonary fibrosis in one lung, the other lung being the site of severe and diffuse emphysema, suggesting that the fibroblastic reticulum may be prevented from expanding because of underlying emphysema.
AE was observed in 46.9% of patients with AIPF, which was more frequent than in controls with symmetrical IPF (p=0.004). The prevalence of 17.2% in our control population is very close to that published by Kim et al (9.6% at 2 years using Kondoh's criteria for AE but 18% using Akira's criteria).19 According to a new proposed international definition, only cases with bilateral abnormalities on HRCT scanning should be termed ‘acute exacerbation’.2 Two-thirds of patients with AIPF experienced virtually unilateral episodes but fulfilled Akira's criteria for AE.4 AE developed in the most fibrotic lung and, after recovery, ground glass opacities were replaced by reticulations. These findings support the hypothesis of AE as a sudden deterioration in the primary disease process rather than a distinct additional event. Again, they point to the role of GER. The aspiration of gastric contents is a well-known cause of diffuse alveolar damage,20 and acute worsening of IPF may be provoked by a particularly severe episode of reflux, as suggested in exacerbations of chronic obstructive pulmonary disease.21 Although primarily unilateral in most of our cases with AIPF, AE was fatal only when bilateral, probably due to the respiratory reserve provided by the least affected lung. This is in keeping with the study by Akira et al which showed that patients with IPF who die of a second episode of AE show a significant extension of opacities from a peripheral pattern to a more diffuse pattern.22
In patients with IPF the extent of fibrosis on the HRCT scan is strongly correlated with Tlco.5 23 In contrast, Tlco was a poor indicator of disease severity in AIPF, as judged by the lack of association between Tlco and the global fibrosis score. Furthermore, despite a similar global fibrosis score and FVC, patients with AIPF had a significantly higher Tlco compared with controls. This difference in Tlco probably produces a different mode of fibrosis progression and distribution in the two groups. It is well known that Tlco reflects whole lung involvement but is much more influenced by base involvement because of physiological ventilation/perfusion regional inhomogeneity.24 In patients with symmetrical IPF, pulmonary fibrosis usually progresses simultaneously from the base to the top of the two lungs. Both bases are equally affected. In those with AIPF, pulmonary fibrosis also progresses from the base to the top of the more affected lung before involving the other one, so global extension of pulmonary fibrosis is similar but one of the two bases is spared for relatively longer.
Surprisingly, patients with AIPF were significantly older than controls, which is difficult to explain. It is possible that IPF is detected later in these patients because of a less symptomatic disease.
Our study has some limitations. As a retrospective case series study it was not designed to evaluate the prevalence of AIPF. However, the French ongoing prospective cohort study on IPF provides an estimation of its incidence.25 Included patients were systematically scored and the same ratio was used to define asymmetry. Ten of the 99 patients (10.1%) first included had a significant asymmetry on the HRCT scan at inclusion. Moreover, there may be a bias in the estimation of GER between patients with AIPF and controls as more attention is given by clinicians to search for GER in patients with an asymmetrical form of the disease. Finally, our population was small and the study was purely descriptive so no definite conclusion can be reached on the pathogenesis of AIPF.
In conclusion, AIPF may be favoured by several underlying conditions including, but not limited to, GER. GER may play a role both in driving the insidious progression of IPF and in promoting AE. AIPF also gives new insight into the key role of locoregional factors in the expansion of pulmonary fibrosis. Tlco may not be adapted to evaluate the severity of this particular form of IPF. Further studies are needed to assess more clearly the frequency and specific risk factors for AIPF and to determine the real impact of this form of IPF on clinical practice.
References
Footnotes
See Editorial, p 183
Linked articles 149096.
Competing interests DV has received consultancy fees from Intermune, Actelion and Sanofi (less than $4000 in the last 5 years) and expenses from AstraZenica, Glaxo, Boehringer and Nycomed for travel and accommodation at ATS, ERS and CPLF meetings.
Ethics approval This study was conducted with the approval of the institutional review board.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Airwaves