Article Text

Abstract
Rationale Traditional genome-wide association studies (GWASs) of large cohorts of subjects with chronic obstructive pulmonary disease (COPD) have successfully identified novel candidate genes, but several other plausible loci do not meet strict criteria for genome-wide significance after correction for multiple testing.
Objectives The authors hypothesise that by applying unbiased weights derived from unique populations we can identify additional COPD susceptibility loci.
Methods The authors performed a homozygosity haplotype analysis on a group of subjects with and without COPD to identify regions of conserved homozygosity haplotype (RCHHs). Weights were constructed based on the frequency of these RCHHs in case versus controls, and used to adjust the p values from a large collaborative GWAS of COPD.
Results The authors identified 2318 RCHHs, of which 576 were significantly (p<0.05) over-represented in cases. After applying the weights constructed from these regions to a collaborative GWAS of COPD, the authors identified two single nucleotide polymorphisms (SNPs) in a novel gene (fibroblast growth factor-7 (FGF7)) that gained genome-wide significance by the false discovery rate method. In a follow-up analysis, both SNPs (rs12591300 and rs4480740) were significantly associated with COPD in an independent population (combined p values of 7.9E–7 and 2.8E–6, respectively). In another independent population, increased lung tissue FGF7 expression was associated with worse measures of lung function.
Conclusion Weights constructed from a homozygosity haplotype analysis of an isolated population successfully identify novel genetic associations from a GWAS on a separate population. This method can be used to identify promising candidate genes that fail to meet strict correction for multiple testing.
Statistics from Altmetric.com
Key messages
What is the key question?
Can information from isolated populations improve our ability to detect novel genetic variants in genome-wide association studies (GWASs)?
What is the bottom line?
We identified statistically significant polymorphisms in a novel chronic obstructive pulmonary disease (COPD) gene (FGF7), which we replicated in an independent population.
Why read on?
We demonstrate the use of a novel method (homozygosity haplotype analysis) for identifying genomic regions that are inherited from a common ancestor, and use this information to weight a GWAS of COPD to identify novel genetic variants that are associated with increased risk of disease.
Introduction
Traditional genome-wide association studies (GWASs) have identified novel susceptibility loci for complex diseases such as chronic obstructive pulmonary disease (COPD).1–3 Because the effect size of most common disease-susceptibility variants is modest, GWASs of complex diseases require large sample sizes to achieve statistically significant results after correction for multiple testing. Weighting the results of GWASs according to prior information (eg, from linkage studies) may significantly improve the power to detect associations that do not meet genome-wide (GW) significance.4
Homozygosity mapping is a promising technique to identifying regions of the genome that are more likely to contain disease-susceptibility loci. Although initially developed to identify rare susceptibility mutations for monogenic traits in families,5 homozygosity mapping has recently been successfully applied to the study of complex diseases.6 7 While techniques vary, the concept underlying all homozygosity haplotype (HH) methods is that regions of homozygosity are more likely to contain disease-susceptibility loci in affected subjects than in unaffected individuals.8
Using high-density single nucleotide polymorphism (SNP) arrays, Miyazawa et al developed a novel variation of homozygosity mapping that tests whether multiple subjects share the same genotype among homozygous SNPs, and then constructed a region of conserved homozygosity haplotype (RCHH) that reflects the transmission of the haplotype from a founder population. In theoretical simulations, this method was shown to be a viable method to detect disease-susceptibility loci in recently admixed populations.9 We hypothesised that application of this method to a genetic isolate in Costa Rica would result in detection of an over-representation of regions of conserved homozygosity in subjects affected with COPD compared with unaffected subjects. In this report, we first identify regions of conserved homozygosity in Costa Ricans and then show that weights derived from these regions can be applied to GWASs in non-isolated populations to identify novel disease-susceptibility loci for COPD. Using this approach, we identify a novel COPD candidate gene (fibroblast growth factor-7 (FGF7)).
Materials and methods
Study population
The primary study population consisted of 58 subjects with COPD (cases) and 57 subjects without COPD (controls) in the Genetic Epidemiology of COPD in Costa Rica study. Cases were recruited from patients attending four adult hospitals in San José (Costa Rica) and their affiliated clinics, and through newspaper advertisements. Control subjects were recruited from individuals attending a smoking-cessation clinic at the Institute for Pharmaco-dependency in San José, and through newspaper advertisements. To ensure their descent from the founder population of the Central Valley of Costa Rica (which is predominantly of Spanish and Native American ancestry), all participants were required to have at least six great-grandparents born in the Central Valley. Additional inclusion criteria for cases were ages 21–71 years, physician-diagnosed COPD, ≥10 pack-years of cigarette smoking, a forced expiratory volume in one second (FEV1) ≤65% predicted and an FEV1/forced vital capacity (FVC) ratio of ≤70% after bronchodilator administration (180 μg of albuterol by metered dose inhaler). Controls were recruited on the basis of the same criteria for age and smoking history, but they had to have no physician-diagnosed COPD and normal spirometry. Exclusion criteria for cases and controls included history of chronic pre-existing chronic lung disease (eg, bronchiectasis) and severe α-1-antitrypsin deficiency (for cases), based on molecular phenotyping. The baseline characteristics of this cohort are in listed in the online supplementary table 2.
Written consent was obtained from participating subjects. The study was approved by the institutional review boards of the Hospital Nacional de Niños (San José, Costa Rica), Partners Healthcare System (Boston, Massachusetts, USA), and participating National Emphysema Treatment Trial (NETT), Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) and Norway centres.
Genotyping of Costa Rican cohort
High-density SNP genotyping was performed using the Illumina Quad 610 platform at the Channing Laboratory, Boston, Massachusetts, USA. Cases and controls were randomly distributed among batches, and each batch contained a replicate sample. All subjects had an SNP call rate >95%. After quality control measures (see online supplementary table 1), a total of 558 929 SNPs were acceptable for analysis.
Collaborative COPD cohorts for the primary GWAS
Three populations with a total of 2940 cases and 1380 controls were used for the primary GWAS: (1) subjects in a case–control study of COPD in Norway (838 cases and 791 controls)3; (2) subjects in the NETT (366 cases) and the Normative Aging Study (414 controls)10 11 and (3) 1736 cases and 175 controls from the multicentre ECLIPSE study.12 All controls were current or former smokers with normal spirometry, and all cases with COPD had moderate to very severe disease according to the Global Initiative for Chronic Obstructive Lung Disease classification.13
Lovelace Smokers Cohort
The top SNPs in novel genes were replicated in a cohort of 1845 smoking adults in New Mexico, 424 (23%) of whom were classified with COPD based on an FEV1/FVC ratio below the fifth percentile of the predicted value, also referred to as the lower limit of normal.14 Of the 1845 participants, 1411 (77%) were Caucasian and 313 (17%) were Hispanic. The protocols for subject recruitment and data collection for the Lovelace Smokers Cohort have been previously described in detail.15 The two SNPs (rs12591300 and rs4480740) were genotyped by allelic discrimination using Taqman assay (Applied Biosystems, Foster City, California, USA). The case–control association analysis was first performed in all subjects, and then separately in Caucasians and Hispanics. All analyses were adjusted for age, gender and pack-years of cigarette smoking; the analysis of all subjects was additionally adjusted for self-declared ethnicity.
Gene expression analysis
For the top novel candidate genes, we examined the correlation of gene expression in lung tissue with COPD intermediate phenotypes (FEV1 and FEV1/FVC ratio) in a previously published COPD biomarker discovery study.16 This cohort consists of 56 subjects with varying degrees of obstruction who underwent lung resection for a solitary pulmonary nodule. RNA expression profiling was completed using the Affymetrix U133 Plus 2.0 array, as previously described.16 Expression correlation with quantitative phenotypes was conducted as previously described.16
Statistical analysis
Construction of RCHHs
RCHHs were identified using the method described by Miyazawa et al.9 In brief, for any given individual all heterozygous SNPs were ignored and the SNP location was scored with the value of the allele for that subject. Subjects are compared only across SNPs that are scored. RCHHs are defined by runs of SNPs that share the same allele at the homozygous locations across multiple subjects, ignoring heterozygous SNPs. The size of the shared segments between any two individuals was set at 3.0 cM (roughly and approximately three million base pairs), which in theoretical work conducted by Miyazawa et al9 reduced the false positive and false negative rates of discovery. A theoretical ancestral segment was then constructed from the largest subgroup of subjects sharing a particular RCHH (see online supplementary figure 1). While any two subjects must have at least 3.0 cM of sharing, the size may be much smaller when comparing across multiple subjects (online supplementary figure 2). If more than one ancestral region is identified at a particular chromosomal location, the region shared by the most number of subjects is used (online supplementary figure 3). The total number of cases and controls sharing this ancestral allele is used to calculate a p value based on a standard normal distribution.
For the primary analysis of the collaborative COPD cohort, logistic regression analysis was performed under an additive genetic model for each SNP, adjusting for age, pack-years of smoking and the first 16 principal components (to adjust for population stratification). The p values from all RCHHs identified in Costa Rica were then used to construct a cumulative weight for each SNP from the recent GWAS of COPD in the combined cohort of Norway, ECLIPSE and NETT–Normative Aging Study using the method developed by Roeder et al.4 Briefly, the weighting method utilises prior information (in this case, the p value representing the degree of over-representation of a region of the genome in cases versus controls) to upweight or downweight p values from an association study (in this case, the GWAS of COPD in the collaborative cohort). In order to maintain an overall α level of 0.05, the assigned weights across the genome average to 1. For this study, SNPs that did not fall inside of an RCHH (and therefore did not have a p value) were assigned a p value of 1 (and therefore a weight approaching zero). This is a more conservative approach than excluding these SNPs from consideration. The method then calculates a false-discovery rate (FDR) using the method described by Benjamini and Hochberg17 to correct for multiple testing.
The RCHHs were created and compared with HHAnalysis (available at http://www.hhanalysis.com). Association analysis was performed using PLINK V.1.07 (http://pngu.mgh.harvard.edu/purcell/plink). The weighting procedure was performed using software developed by Roeder et al4 (http://wpicr.wpic.pitt.edu/wpiccompgen/). All other statistical analysis was performed using R V.2.9.0 (http://www.R-project.org).
Results
Identification of RCHHs in Costa Rica and construction of weights
In total, 2318 RCHHs were identified in the Costa Rican cohort. Of these 2318 regions, 576 were significantly (p<0.05) over-represented in cases compared with controls; none of the regions were significantly more frequent in controls than cases. The median size of the significant regions was 105 kb, and the largest was 7.2 Mb. Online supplementary table 3 shows the top 20 p values representing 100 RCHHs in Costa Rica.
Each SNP in the combined collaborative COPD cohort was then mapped to an RCHH and assigned the p value of the whole region. SNPs that did not map to an RCHH were assigned a p value of 1. The mapped p values across all genotyped SNPs were then used to create weights using a cumulative distribution function. The algorithm is constructed so that the mean weight across all SNPs is 1: some SNPs are upweighted and a much larger fraction is downweighted. The nominal p value is divided by the weight to obtain the weighted p value.
Application of weights to the COPD GWAS
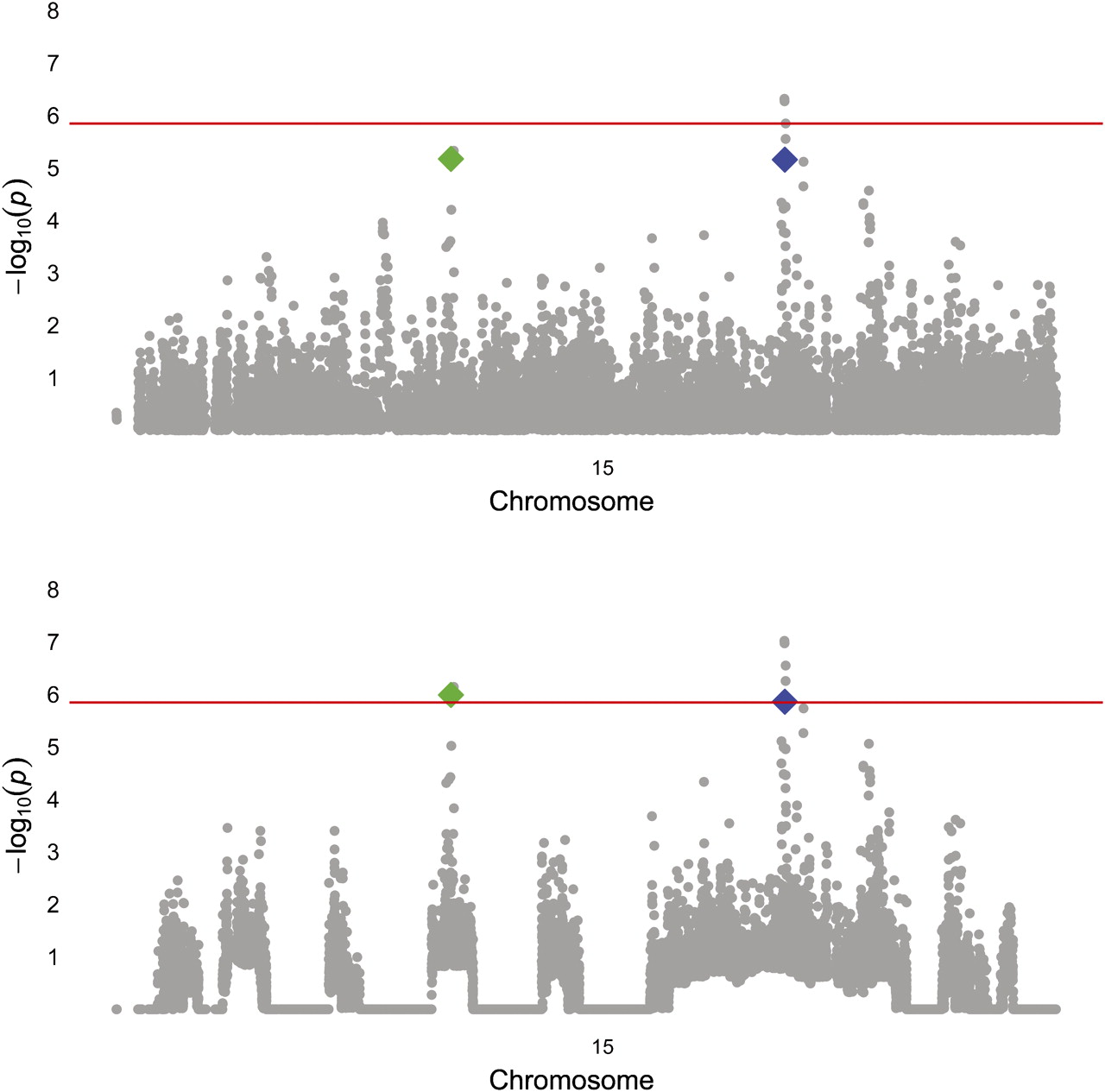
We applied the weights derived from the HH analysis above to reanalyse GW genotypic data in a cohort of subjects of European descent that was previously employed for a traditional GWAS of COPD. After weighting, 14 SNPs were significant at an FDR-corrected α of 0.05. The top five SNPs from the unweighted GWAS retained their original ranks, but several SNPs that did not achieve GW significance in the traditional GW association analysis became more statistically significant and moved higher in the list (table 1). Of these SNPs, those in the gene for FAM13A were identified in the original analysis of the GWAS,1 and SNPs in IREB218 and CHRNA33 have been implicated in COPD affection status in prior candidate-gene and GWASs. Two of the other SNPs lie in two novel candidate genes for COPD, FGF7 and proteasome subunit, α-type, 4 (PSMA4) (figure 1). The RCHH in Costa Rica that contains FGF7 was present in seven cases and no controls, and the RCHH containing PSMA4 was present in five cases and no controls.
FDR significant* results from weighted GWAS

{kind=link}
Manhattan plot of chromosome 15, before (top) and after weighting. rs4480740 (Green) is in the gene FGF7 and rs2036534 (blue) is in the promoter of PSMA4. The red line indicates the FDR corrected α level for genome-wide significance. FDR, false-discovery rate; FGF7, fibroblast growth factor-7; PSMA4, proteasome subunit, α-type, 4.
The regions containing the genes CHRNA3 and IREB2 were also over-represented in cases compared with controls (p<0.05), and after weighting they were GW significant by FDR. While there was an RCHH containing FAM13A identified in the Costa Rican cohort, it was only seen in one case and no controls.
Replication in Lovelace Smokers Cohort
The top two SNPs in or near FGF7 were genotyped in the 1845 smoking adults in the Lovelace Smokers Cohort. The minor alleles of both SNPs conferred increased odds for COPD in the whole population in the same direction as the original collaborative COPD cohort (table 2). Among the Hispanic subgroup, the effect size was larger and in the same direction for both SNPs, but only rs12591300 showed a significant association with COPD affection status.
Combined p values for replication of FGF7 SNPs
Gene expression analysis
Our previous studies indicate that gene expression patterns associated with quantitative, intermediate COPD phenotypes are most informative for the discovery of disease-associated genes.16 18 19 We examined disease-associated expression patterns for our novel candidate genes in a previously published GW expression data set from 56 subjects with varying degrees of airflow obstruction (assessed by spirometric measures of lung function (FEV1 and FEV1/FVC ratio)).16 Expression of FGF7 (as defined by multiple and independent probe sets) was significantly negatively correlated with both FEV1 (nominal p value <0.01) and FEV1/FVC ratio (nominal p value <0.01), indicating increased expression associated with increased disease severity. Expression in COPD subjects was increased compared with control subjects, but the difference was not statistically significant. PSMA4 expression was not correlated with lung function and was not differentially expressed in cases versus controls.
Discussion
While successful in identifying novel candidate genes, GWASs of complex traits are unlikely to identify all potential common disease-susceptibility variants because of limited power if strict criteria for GW significance are applied. In the absence of a very large sample size, novel methods are needed to identify disease-susceptibility variants not meeting GW significance. We identified RCHHs for COPD in a GW case–control study in Costa Rica. After applying a weighting method based on the degree of significance of these regions to a GWAS of COPD cases and controls of European descent, we identified two SNPs in a novel candidate gene for COPD (FGF7) and demonstrated that several SNPs in the previously identified candidate genes IREB2 and CHRNA3 met GW criteria for statistical significance. An SNP in another novel gene (PSMA4) was GW significant after weighting. However, expression of PSMA4 in the lung was not associated with COPD phenotypes, and thus the observed association is likely due to linkage disequilibrium with the nearby genes CHRNA3 and IREB2. We then replicated the two FGF7 SNPs in an independent cohort of smoking adults, and showed that they are both significantly associated in the same direction with COPD. Notably, the effect sizes in Hispanics are larger than in the overall cohort, suggesting that these alleles confer greater risk in this population. This Hispanic population in New Mexico has a similar proportion of European and Native American ancestry as the Costa Rican cohort,20 21 so another likely possibility is that patterns of linkage disequilibrium may be different between Hispanics and Caucasians in this genomic region, and that these SNPs are tagging a haplotype or functional SNP in the Hispanic subjects. Additionally, there was a trend towards increased lung tissue expression of FGF7 in an independent cohort of COPD subjects, in whom there was a significant negative correlation between FGF7 expression and FEV1 and FEV1/FVC ratio.
There are several plausible methods for weighting chromosomal regions in GWAS, including upweighting previously identified candidate genes, coding variants, exons and promoter regions. However, these weighting strategies work counter to one of the strengths of a GWAS: its hypothesis-free nature. Using HHs as a weighting method avoids the pitfall of these other weighting strategies because they are constructed using a hypothesis-free method, so the weights are unbiased with respect to prior knowledge.
One of the main strengths of our study is that it shows the power of using HH analysis in an isolated population to investigate common diseases. While our sample size was small, Miyazawa et al9 have previously shown in simulated data that HH analysis has the ability to identify the region containing an SNP inherited identity-by-descent from a distant common ancestor using only 45 cases and 45 controls. In our own data, we were able to show that the previously identified candidate genes IREB2 and CHRNA3 fall within an RCHH that is significantly over-represented in subjects with COPD. When combined with results from a weighted GWAS in an independent cohort with adequate sample size, we were able to show that variants in these genes are significant after correction for multiple testing.
Two novel genes are contained within significant regions of conserved homozygosity, and after weighting they are significant by FDR correction. The first, FGF7, was identified in cultured human embryonic lung fibrobasts,22 and plays a role in promoting wound healing23 and protecting airway epithelium from oxidant injury in mice.24 One of the SNPs identified in this study (rs4480740) is in an intron of FGF7, and the other (rs12591300) lies immediately upstream of FGF7 in an intron of hypothetical protein LOC196951. In a GWAS of FEV1 in the British 1958 Birth Cohort, five out of the nine SNPs genotyped in FGF7 were significantly (p<0.05) associated with differences in lung function, although not the two SNPs identified in this study.25 FGF7 has been shown to protect against oxidative stress response specifically in the lung epithelium,24 so increases in expression associated with disease progression may indicate a greater burden of injury. A limitation of our study is the lack of experimental evidence for an effect(s) of the SNPs identified in FGF7 on gene expression. We hypothesise that these SNPs cause decreased expression of FGF7, which could affect antioxidant mechanisms protecting against detrimental effects of cigarette smoking on the lung. Alternatively, FGF7 may play a role in disease susceptibility through its role in epithelial development during embryogenesis by influencing epithelial responses to cigarette smoke. Since it is unclear whether increased FGF7 expression is a marker of exposure to oxidant injury or a cause of epithelial damage, further work must be done to characterise the role of these SNPs on FGF7 expression.
The HHAnalysis algorithm works best under certain assumptions, namely that (1) the risk alleles were introduced into the population from a population of common ancestors within the last several hundred years, (2) the target population is genetically isolated, (3) the number of common ancestors introducing the risk allele is small and that (4) the risk of the disease allele is moderate to high. Violations of these assumptions reduce the theoretical expected size of the RCHH and/or the association of the RCHH with disease, which reduces the power of the algorithm to detect them. Genetic and historical data for the population of the Central Valley of Costa Rica suggest that the first three assumptions are met. As in most association studies of complex disease, the effect size of a risk allele is likely small to moderate at most, and we expect that this has somewhat reduced our power.
Whereas other homozygosity mapping methods are primarily designed to detect recessive alleles, the HHAnalysis method instead uses homozygosity to identify ancestral regions inherited from a common ancestor. These regions from a common ancestor can harbour risk alleles that operate under recessive, dominant or additive models. However, the HHAnalysis algorithm would also detect copy number variation that results in the deletion of a single allele. While this may explain a fraction of the regions identified, the top novel SNPs identified in FGF7 do not fall within known regions of copy number variation according to the Database of Genomic Variants.26
In summary, we have shown that weights obtained from HH analysis in an isolated population can improve the power to detect novel variants in GWAS in non-isolates. In addition to confirming results for previously identified variants in IREB2 and CHRNA3, we have identified variants in a novel candidate gene (FGF7) for COPD. The validity of this gene is supported by replication in an independent cohort of smoking adults, and expression data showing consistent and significant patterns associated with COPD intermediate lung function phenotypes. Further analysis of these genes in the Costa Rican cohort and functional studies should yield insights into the causative SNPs or haplotypes that underlie the associations identified in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding The Genetic Epidemiology of COPD in Costa Rica is supported by grant R01HL073373 from the National Heart, Lung, and Blood Institute. The National Emphysema Treatment Trial (NETT) is supported by contracts with the National Heart, Lung, and Blood Institute (N01HR76101, N01HR76102, N01HR76103, N01HR76104, N01HR76105, N01HR76106, N01HR76107, N01HR76108, N01HR76109, N01HR76110, N01HR76111, N01HR76112, N01HR76113, N01HR76114, N01HR76115, N01HR76116, N01HR76118, N01HR76119), the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality (AHRQ). The Norway cohort and the ECLIPSE study (http://clinicaltrials.gov identifier NCT00292552; GSK Code SCO104960) are funded by GlaxoSmithKline. The Lovelace Smokers Cohort is supported by funding from the State of New Mexico (appropriation from the Tobacco Settlement Fund) and by grant RO1 ES015482 from the National Institute of Environmental Health Sciences. We acknowledge use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02.
Competing interests None.
Ethics approval Institutional Review Board of University of Pittsburgh, Partners Health Care (Boston), participating NETT centres, Boston VA, Norway, Costa Rica, and Lovelace Respiratory Institute.
Provenance and peer review Not commissioned; externally peer reviewed.