Article Text

Abstract
Background The well-characterised airway inflammatory phenotypes of asthma include eosinophilic, neutrophilic, mixed eosinophilic/neutrophilic and paucigranulocytic asthma, identified based on the proportion of sputum granulocytes. Systemic inflammation is now recognised as an important part of some airway diseases, but the involvement of systemic inflammation in the pathogenesis of airway inflammatory phenotypes of asthma remains unknown.
Methods Induced sputum samples and peripheral blood were collected from participants with asthma (n=36). Airway inflammatory cell counts were performed from induced sputum and inflammatory phenotype assigned based on the airway eosinophil and neutrophil cut-offs of 3% and 61%, respectively. RNA was extracted from whole blood and gene expression profiles were generated (Illumina Humanref-8 V3) and analysed using GeneSpring GX11.
Results There were six genes classified as differentially expressed between the four asthma phenotypes, including the α-defensins (DEFA) 1, 1B, 3 and 4 and neutrophil proteases cathepsin G (CTSG) and elastase (ELA2). Systemic expression of DEFA1,1B,3,4,CTSG and ELA2 was significantly higher in the neutrophilic asthma (NA) phenotype. Microarray results of the α-defensins and neutrophil proteases were successfully validated using real-time PCR. Plasma elastase was significantly increased in people with neutrophilic airway inflammation.
Conclusion There is systemic upregulation of α-defensin and neutrophil protease expression in NA, which may represent proinflammatory effects on the bone marrow and the release of immature neutrophils into the circulation. This demonstrates a systemic inflammatory component in NA that further differentiates this from other asthma phenotypes and indicates different mechanisms in NA.
- Asthma phenotypes
- systemic inflammation
- gene expression
- defensins
- proteases
- asthma mechanisms
- innate immunity
- neutrophil biology
Statistics from Altmetric.com
- Asthma phenotypes
- systemic inflammation
- gene expression
- defensins
- proteases
- asthma mechanisms
- innate immunity
- neutrophil biology
Key messages
What is the key question?
Are systemic gene expression profiles different between inflammatory phenotypes of asthma?
What is the bottom line?
The neutrophilic phenotype of asthma is associated with increased systemic gene expression of neutrophil α-defensins and proteases elastase and cathepsin G.
Why read on?
This study explores the relationship between systemic gene expression, airway inflammation and asthma phenotypes, and discovers novel systemic markers of neutrophilic airway inflammation in asthma.
Introduction
Asthma is now well recognised to be a heterogeneous disease, whether assessed using clinical factors, causative triggers, response to treatments or types of inflammation present.1–3 The measurement of cellular patterns of inflammation present in induced sputum samples has proved to be useful in identifying inflammatory phenotypes of asthma. We have previously described four distinct cellular subtypes of asthma based on the presence or absence of sputum granulocytes—namely, eosinophilic asthma (EA), neutrophilic asthma (NA), mixed eosinophilic and neutrophilic asthma (ME/NA) and paucigranulocytic asthma (PGA).3 Nadif et al4 have recently reported the presence of similar inflammatory phenotypes of asthma based on the inflammatory cell numbers present in peripheral blood, suggesting there may also be systemic manifestations of the airway inflammatory response in asthma.
The underlying biology of asthma inflammatory phenotypes has not been fully characterised. A Th2-mediated defect forms part of EA, and it is thought that dysregulation of the innate immune response involving neutrophils is important in the pathogenesis of NA.5 6 We have previously shown that NA and EA can be identified by distinct gene expression profiles in peripheral blood neutrophils, and that enhanced systemic neutrophil migration and survival may be involved in the pathogenesis of NA.5 In addition to identifying a neutrophil defect in asthma, these studies also suggested that there may be systemic inflammation in asthma. In other airway diseases such as chronic obstructive pulmonary disease, low-grade systemic inflammation is recognised as a key pathogenic feature of the disease.7
However, it is unclear if there is a relationship between airway and systemic inflammation in asthma, and whether systemic inflammation is related to certain inflammatory phenotypes of asthma. It is thought that airway inflammation can ‘spillover’ into the circulation and this can produce low-grade systemic inflammation.8 However, this concept is challenged by several studies in chronic obstructive pulmonary disease that found no correlation between mediator levels in the sputum and plasma,9 suggesting that pulmonary and systemic responses may be modulated separately. Furthermore, the range of markers used to characterise systemic inflammation has been limited predominantly to C reactive protein and fibrinogen. Better characterisation of systemic pathways involved in asthma may identify potentially important mechanisms. This project investigated gene expression profiles of the inflammatory cells of the blood and related these to inflammatory phenotypes of asthma defined by airway inflammation.
Methods
Participants
Adults with stable asthma were recruited from the John Hunter Hospital Asthma Clinic, NSW, Australia. Asthma was diagnosed according to American Thoracic Society guidelines based upon current (past 12 months) episodic respiratory symptoms, doctor's diagnosis (ever) and demonstrated evidence of airway hyper-responsiveness to hypertonic saline. Exclusion criteria included recent (past month) respiratory tract infection, recent asthma exacerbation, recent unstable asthma or change in maintenance therapy and current smoking (or past smoking within 6 months of cessation). All participants gave written informed consent prior to their inclusion in the study and the Hunter New England Area Health Service and The University of Newcastle Research ethics committees approved the study.
Sputum induction and analysis
Spirometry (KoKo PD Instrumentation, Louisville, Colorado, USA) and sputum induction with hypertonic saline (4.5%) were performed. A fixed sputum induction time of 15 min was used for all participants. For inflammatory cell counts, selected sputum (sputum portions separated from saliva) was dispersed using dithiothreitol. The suspension was filtered and a total cell count of leucocytes and cell viability was performed. Cytospins were prepared, stained (May–Grunwald Giemsa) and a differential cell count obtained from 400 non-squamous cells.
Whole genome gene expression microarrays
Four millilitres of blood was collected by venepuncture and subjected to red blood cell lysis. The remaining white blood cells were stored in buffer RLT (Qiagen, Hilden, Germany) at −80°C until RNA extraction. RNA was extracted using the RNeasy Mini Kit (Qiagen) and quantitated using the Quant-iT RiboGreen RNA Quantitation Assay Kit (Molecular Probes Inc, Invitrogen, Eugene, Oregon, USA). Fluorescence was measured at wavelengths of 485 nm (excitation) and 520 nm (emission) (FLUOstar Optima, BMG Labtech, Victoria, Australia). 500 ng of RNA was reverse-transcribed into cRNA and biotin-UTP labelled using the Illumina TotalPrep RNA Amplification Kit (Ambion, Texas, USA). 750 ng cRNA was hybridised to the Illumina Sentrix HumanRef-8 Version 3 Expression BeadChips (Illumina, San Diego, California, USA) using standard protocols. Each BeadChip measured the expression of 24 590 probe sets and was scanned using the Illumina Bead Station and captured using BeadScan 3.5.11 (Illumina).
Quantitative real-time PCR validation
Peripheral blood RNA (200 ng) was reverse-transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, California, USA). Taqman qPCR primer and probes for DEFA1, DEFA1B and DEFA3 (Hs00234383_m1), DEFA4 (Hs00157252_m1), CTSG (Hs00175195_m1) and ELA2 (Hs00357734_m1) were purchased in kit form from Applied Biosystems. The same primer/probe detected the expression of DEFA1, DEFA1B and DEFA3 owing to the high homology between the genes. PCR primers and probes were combined with Taqman gene expression master mix according to the manufacturer's instructions in Singleplex real-time PCRs (7500 Real-Time PCR System, Applied Biosystems). The results were calculated using 2−ΔΔCt relative to both the housekeeping gene (β-actin) and the mean ΔCt of all samples.
Quantification of protein levels of plasma neutrophil elastase
Levels of active neutrophil elastase (NE) were measured in corresponding available plasma samples (n=26) using the Innozyme Human Neutrophil Elastase Immunocapture Activity Assay Kit (Calbiochem-EMD4biosciences, San Diego, California, USA) according to the manufacturer's instructions. The standard curve used to detect plasma NE activity ranged from 31.25 pg/ml to 2000 pg/ml; samples were run in duplicate and the fluorescence was measured at 360 nm (excitation) and 440 nm (emission).
Asthma subtype classification
The granulocyte cut-off values used were 3% for sputum eosinophils and 61% for sputum neutrophils.3 10 Participants with a sputum eosinophil count of >3% alone were classified as having EA, those with sputum eosinophils >3% and sputum neutrophils >61% were classified as having ME/NA, participants with sputum neutrophils >61% and sputum eosinophils <3% were classified as having NA and those with sputum eosinophils <3% and sputum neutrophils <61% were classified as having PGA.
Statistical analysis
Clinical and cell count data were analysed using Stata 9 (Stata Corporation, College Station, Texas, USA) and reported as mean (SD) for normally distributed data and median (Q1, Q3) for non-parametric data. Statistical comparisons were performed using the multiple comparison ANOVA test for normally distributed parametric data (p<0.05 considered significant) with post-hoc tests between the four groups adjusting for multiple comparisons using the Bonferroni method (p<0.013). The Kruskal–Wallis test was applied to non-parametric data (p<0.05 considered significant) with the post-hoc Kruskal–Wallis test adjusted for multiple comparisons between groups (p<0.004).
Whole genome gene expression data were exported using Genome Studio (Illumina) and analysed using GeneSpring GX11 (Agilent Technologies, Santa Clara, USA). Data were log-transformed, normalised and baseline converted to the median of all samples. Data were filtered and only genes flagged as present (p<0.05 detection value) in all 36 samples were included in the further analysis. Differential gene expression between asthma inflammatory phenotypes was determined using ANOVA with Tukey HSD post-hoc testing (p<0.01) and fold change >2.
Results
Clinical features and inflammatory cells
Clinical details and induced sputum inflammatory cell counts are shown in table 1. Participants with EA had significantly higher rates of atopy than those with NA and significantly higher levels of exhaled nitric oxide than those with NA and PGA. As expected, the percentages of sputum granulocytes and macrophages differed significantly between groups. However, participants with NA had significantly higher absolute numbers of sputum neutrophils, whereas those with ME/NA had an increased percentage of neutrophils but did not have significantly higher absolute numbers of sputum neutrophils than those with EA and PGA. Participants with EA and ME/NA also had significantly higher numbers of sputum eosinophils compared with the other groups, and the level of eosinophilic inflammation was further increased in EA compared with ME/NA.
Clinical characteristics and sputum cell counts of inflammatory phenotypes of asthma
Whole genome gene expression
Whole genome gene expression data (24 526 entities) were filtered and only entities flagged as present (p<0.05 detection value) in all 36 samples were included in further analysis (17 004 entities). There were 175 genes that were significantly different (p<0.01) in expression between the four asthma groups. Only six of these genes were differentially expressed at a fold change of >2 in at least one comparison. The six genes differentially expressed between the four asthma phenotypes were the α-defensins (DEFA1, DEFA1B, DEFA3 and DEFA4) and the neutrophil proteases cathepsin G (CTSG) and elastase (ELA2). Gene expression of DEFA1, DEFA1B, DEFA3, DEFA4, CTSG and ELA2 was significantly upregulated in NA (figure 1, table 2).

Systemic upregulation of the α defensins DEFA1 (A), DEFA1B (B), DEFA3 (C), DEFA4 (D) and the neutrophil proteases cathepsin G (E) and elastase (F) in neutrophilic asthma. Microarray data are log-transformed and normalised to the median of all samples. The bars represent the mean of each group. p<0.01 (Tukey post-hoc test) versus *EA, #PGA and ∼ME/NA. EA, eosinophilic asthma; ME/NA, mixed eosinophilic and neutrophilic asthma; NA, neutrophilic asthma; PGA, paucigranulocytic asthma.
Systemic upregulation of defensin and neutrophil protease gene expression in neutrophilic asthma
Correlations
Peripheral blood gene expression of DEFA1, DEFA1B, DEFA3, DEFA4, ELA2 and CTSG was significantly correlated with airway neutrophil proportion and number and with each other (table 3).
Pearson correlations between peripheral blood differentially-expressed genes in neutrophilic asthma determined by microarray and airway neutrophil proportion and number
Real-time PCR validation
Microarray results of the α-defensins (DEFA1, DEFA1B, DEFA3, DEFA4) and neutrophil proteases (CTSG, ELA2) were further validated using real-time PCR. Microarray results were highly correlated with the real-time PCR results (using qPCR ΔCt: DEFA1, r=−0.94; DEFA1B, r=−0.90; DEFA3, r=−0.93; DEFA4, r=−0.85; ELA2, r=−0.91; CTSG, r=−0.83). Gene expression of DEFA1, DEFA1B, DEFA3 (p=0.012), DEFA4 (p<0.001), CTSG (p=0.003) and ELA2 (p<0.001) were significantly increased in NA (figure 2). The fold changes observed were generally larger with real-time PCR than with microarray (figure 3).

Real-time PCR confirmation of the upregulation of α defensins (A, B) and neutrophil proteases (C, D) in NA. The bars represent the mean of each group and the error bars represent±SEM. EA, eosinophilic asthma; ME/NA, mixed eosinophilic and neutrophilic asthma; NA, neutrophilic asthma; PGA, paucigranulocytic asthma.
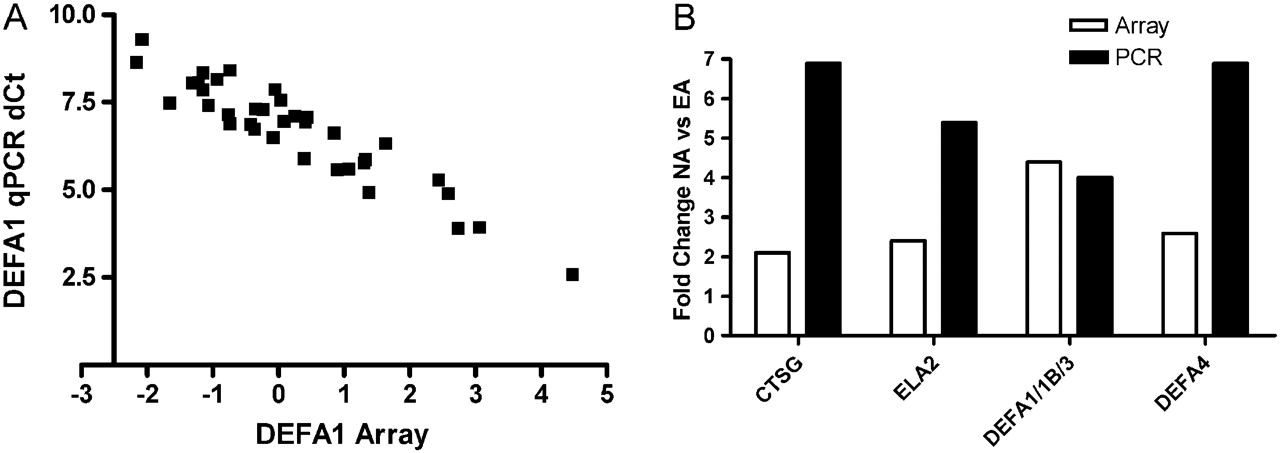
Microarray and real-time PCR results were highly associated. (A) Correlation of normalised microarray and real-time PCR ΔCt data for DEFA1. (B) Fold difference in gene expression between neutrophilic asthma (NA) and eosinophilic asthma (EA) detected were mostly higher using real-time PCR (black bars) than with microarray (white bars).
Plasma neutrophil elastase
Corresponding plasma samples were assayed for active NE where samples were available (n=26/36). NE was not detected in the plasma of participants with PGA (n=7) but it was detected significantly more in participants with NA (n=4/5, 80%; p=0.010); ME/NA (n=6/7, 86%; p=0.005) and EA (n=6/7, 86%; p=0.005). Levels of NE were significantly higher in participants with neutrophilic airway inflammation than in those without neutrophilic airway inflammation (figure 4; p=0.023).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Active plasma neutrophil elastase (pg/ml) was increased in participants with neutrophilic airway inflammation compared with those without neutrophilic airway inflammation.
Discussion
This study used microarray technology to investigate genome-wide gene expression of peripheral blood in relation to airway inflammatory phenotypes of asthma. We report a marked systemic upregulation of α-defensins (DEFA1, DEFA1B, DEFA3 and DEFA4) and neutrophil proteases (ELA2 and CTSG) in NA that was distinct from EA, PGA and ME/NA. The expression of α-defensins and neutrophil proteases significantly correlated with the proportion and number of airway neutrophils, and these results were successfully validated using real-time PCR. Additionally, an increased level of active NE was detected in the plasma of participants with neutrophilic airway inflammation, suggesting systemic neutrophil activation.
The relationship between low-grade systemic inflammation and airway inflammation in asthma remains unknown. We report a significant relationship between the gene expression of α-defensins and neutrophil proteases and the accumulation of neutrophils in the airway. This suggests that there is increased systemic activation and migration of neutrophils to the airways in NA, and that the α-defensins and neutrophil proteases are involved in this process. The accumulation of airway neutrophils is reported to be directly associated with the activation state of circulating neutrophils.11 Airway neutrophil accumulation could therefore be due to an enhanced activation and migration of circulating neutrophils, which is supported by our previous findings of increased expression of genes relating to enhanced cell motility and survival and production of interleukin 8 (IL-8) in peripheral blood neutrophils in non-EA.5 Collectively, these findings identify a specific systemic inflammatory component that is present in NA and not in EA, PGA or ME/NA, and is related to neutrophil primary granules.
An intriguing finding of this study was that ME/NA was similar in systemic gene expression to EA and PGA. This is the first time that a difference between ME/NA and NA has been demonstrated. This suggests that the neutrophilia observed in ME/NA is a local airway response, as opposed to the neutrophilia in NA which has a systemic component involving circulating neutrophil dysfunction. Similarities have previously been found between ME/NA and NA in terms of the level of innate immune mediators such as IL-8 in induced sputum.6 However, our recent sputum gene expression study showed that subjects with ME/NA were predominantly clustered with EA rather than NA.12 Recent work has shown that corticosteroid treatment can induce a sputum neutrophilia,13 raising the possibility that ME/NA represents corticosteroid-overtreated EA.
Innate immunity is dysregulated in NA.6 An important aspect of lung innate immune responses is the production of antimicrobial proteins such as the α- and β-defensins.14 Defensins are small arginine-rich cationic peptides with antimicrobial activity against a broad range of pathogens including bacteria, fungi and some enveloped viruses, and exert this antimicrobial effect through membrane permeabilisation of the microbe.15 Six α-defensins have been identified, including four neutrophil defensins also known as human neutrophil peptides (HNPs 1–4). HNPs 1–4 are predominantly produced by neutrophils, stored in neutrophil azurophilic granules and released in large quantities upon neutrophil activation. The neutrophil defensin genes DEFA1 and DEFA3 are subject to gene copy number variation, which could potentially relate to their level of expression and regulation of immune responses.16 Intratracheal instillation of α-defensins in mice leads to acute lung inflammation and dysfunction involving neutrophil influx and elastase release.17 The α-defensins have a cytotoxic effect, induce IL-8 and IL-1β gene expression, IL-8 protein production and NF-κB binding activity in human bronchial epithelial cells, thereby promoting neutrophilic airway inflammation.18 They also have chemoattractant properties towards immature dendritic cells, T cells and mast cells.19 20 α-defensin-1 can increase mucin gene and protein expression, which may contribute to excess mucus production.21 High levels of α-defensins in airway secretions have been reported in respiratory conditions such as cystic fibrosis,22 idiopathic pulmonary fibrosis and acute respiratory distress syndrome.23 Deficiency of α1-antitrypsin is a condition whereby neutrophil proteases and α-defensins are elevated simultaneously.24 We have previously identified differential expression of proteolytic enzymes in airway samples in asthma phenotypes.25 In that study there was an excess of free NE in airway secretions from subjects with NA, implying insufficient antiprotease activity in NA. An excess of α-defensins and neutrophil serine proteases may therefore underlie the upregulated inflammatory and innate immune response reported in NA, including upregulated proinflammatory IL-8, IL-1β, NF-κB related pathways, and this should be further investigated as a potential cause of NA.
The serine neutrophil proteases cathepsin G and elastase are also found in the azurophilic granules of neutrophils and released upon neutrophil activation. Their primary function is to degrade and kill engulfed microorganisms in the phagolysosome; however, these molecules also participate in extracellular matrix degradation, tissue remodelling, proteolysis of surfactant proteins, activation of matrix metalloproteinases, proteolytic cleavage of cytokines and receptors and cell signalling.26 NE can aggravate inflammation a number of ways, which include increasing the release of IL-8 through stimulation of TLR4,27 stimulating goblet cell degranulation, mucus hypersecretion leading to airflow obstruction.28 29 Plasma levels of NE have not been investigated previously in asthma phenotypes.
The main components of neutrophil azurophilic granules were found to be upregulated in participants with NA in this study. These peptides are synthesised and packaged into granules in the bone marrow in neutrophil precursor cells called promyelocytes.30 Circulating mature neutrophils contain substantial amounts of the formed mature peptides, but are no longer transcribing their mRNA.31 High levels of defensins in marrow neutrophils are associated with a high ratio of immature to mature neutrophilic granulocytes,32 suggesting that the upregulation of defensin and protease mRNA expression in NA may represent the presence of immature neutrophils in the circulation. Under normal conditions granulocyte precursors do not leave the bone marrow. This can change in inflammatory conditions such as infection,33 and neutrophils may skip divisions, shortening their maturation time, and leave the bone marrow prematurely. For example, bacterial endotoxin stimulates the bone marrow and promotes the release of myeloperoxidase-rich neutrophils that sequester in pulmonary microvessels.34 This is particularly relevant as higher levels of airway endotoxin have been detected in NA.6 Collectively, the data in this study identify a systemic component to the inflammatory response in NA that is focused on the primary neutrophilic granule and suggest active bone marrow involvement in the pathogenesis of the significant airway inflammatory changes in NA.
Clinical differences between airway inflammatory phenotypes of asthma included significantly higher rates of atopy in subjects with EA compared with NA and higher levels of exhaled nitric oxide in EA than in NA and PGA. NA has previously been associated with lower rates of atopy,1 4 and subjects with non-atopic asthma have higher levels of C reactive protein.35 This suggests that atopic status, type of airway inflammation and the presence of systemic inflammation are important features distinguishing asthma phenotypes. A past history of smoking is a frequent characteristic of people with asthma, but there was no difference between smoking histories and asthma phenotype in this study and there was no association between smoking history and gene expression of α-defensins or neutrophil proteases. There were significantly higher numbers of neutrophils in NA but not in ME/NA, and significantly higher numbers of eosinophils in both EA and ME/NA. Thus, although subjects with ME/NA had increased proportions of eosinophils and neutrophils, the degree of the neutrophil response was not as high as in NA and the degree of the eosinophil response was not as high as in EA.
Although this study has found important alterations in systemic gene expression in NA that probably represent the molecular mechanisms underlying this disease, there are several limitations that must be considered. First, we used RNA that was extracted from whole blood, representing a heterogeneous population of cells, and therefore groupings may reflect the cell populations present. However, changes in gene expression do not occur without changes in intracellular signal transduction, and whole blood gene expression biomarkers predicting the NA phenotype would be a useful and feasible sample source to guide treatment strategies and monitor active mechanisms. Second, the sample size in the study is relatively small and systemic gene expression should be further investigated in a larger study population. In spite of the limitations, this study has made significant observations that will assist in our understanding of the mechanisms of neutrophilic airway inflammation in asthma.
In summary, there is systemic upregulation of α-defensins and neutrophil proteases in NA. This suggests that NA has a systemic inflammatory component that is significantly different from other asthma phenotypes and may involve proinflammatory effects on the bone marrow and the release of immature neutrophils into the circulation. This study highlights an association between systemic inflammation and NA and also demonstrates a potential use of peripheral blood gene expression of α-defensins and neutrophil proteases to identify NA. This potential application should be further investigated and validated.
Acknowledgments
The authors acknowledge the technical support from Joanne Smart, Hayley Scott, Naomi Fibbens and Lakshitha Gunawardhana.
References
Footnotes
Funding The University of Newcastle funded the project and KJ Baines is funded through the Hunter Medical Research Institute XStrata Coal Asthma Research Fellowship.
Competing interests None.
Ethics approval The Hunter Area Health Service ethics committee and the University of Newcastle research ethics committee approved this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves