Article Text

Statistics from Altmetric.com
The syndrome of combined pulmonary fibrosis and emphysema (CPFE) in adults has not been previously associated with mutations of the surfactant protein-C (SFTPC) gene.
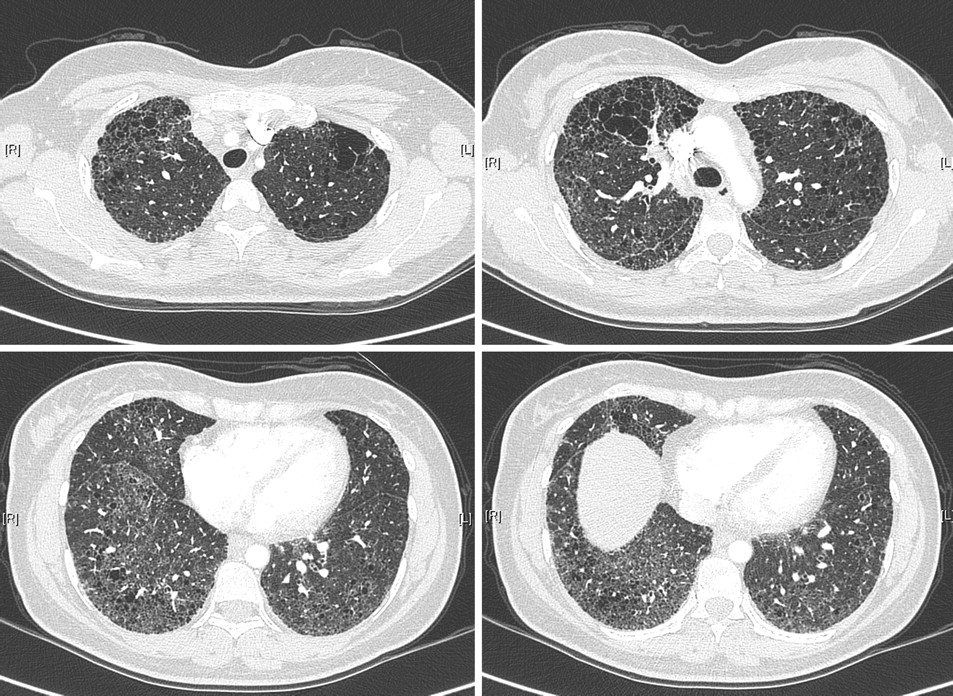
A 32-year-old woman, never smoker, presented with dyspnoea and dry cough, 3 days after a caesarean delivery. Physical examination revealed finger clubbing and bilateral basal crackles. There was no manifestation indicative of connective tissue disease. High-resolution computed tomography (HRCT) of the chest showed conspicuous centrilobular emphysema in the upper zones of the lungs, and diffuse, infiltrative lung disease in the lower zones (figure 1). Emphysema was apparent even in areas devoid of infiltrative changes. The bronchoalveolar lavage differential cell count was 57% neutrophils, 40% macrophages and 3% lymphocytes. Pulmonary function test results 3 months later were forced vital capacity, 62% of predicted value; total lung capacity, 77%; forced expiratory volume in 1 s, 60%; forced expiratory volume in 1 s/vital capacity, 83%; residual volume, 108%; carbon monoxide diffusing factor, 33%; PaO2 on room air, 11.3 kPa; PaO2 after 10-min exercise of 35 W, 7.3 kPa. Video-assisted lung biopsy (see online material) demonstrated disseminated fibrotic thickening of the interalveolar septa, numerous fibroblastic foci, with areas of dense collagen deposition of peribronchiolar predominance, and peribronchiolar emphysema, especially in the upper zones. Echocardiography showed normal heart cavities, with estimated systolic pulmonary arterial pressure of 40 mm Hg. Anti-nuclear antibodies with a nucleolar pattern were further characterised as anti-fibrillarin antibodies. Alpha-1 antitrypsin level was normal. The patient received oral prednisone for 3 months with no improvement. Two years after presentation, chest HRCT and pulmonary function tests had worsened. The patient declined treatment.

{kind=link}
High-resolution computed tomography of the chest of adult non-smoking patient demonstrating centrilobular emphysema in the upper zones of the lungs, and diffuse, infiltrative lung disease in the lower zones, including bilateral reticulation without subpleural predominance, traction bronchiectasis, architectural distortion and ground glass attenuation.
A girl born to this patient was diagnosed to have interstitial lung disease 3 months later (see online material). Her condition improved with oxygen and corticosteroid therapy.
After informed consent was obtained, sequencing of the five translated exons of the SFTPC gene1 from blood samples demonstrated the same heterozygous I73T substitution in both the child and the mother; neither of them had mutation of the SFTPB and ABCA3 genes.
This is the first report of a phenotype of CPFE syndrome in an adult patient carrying a mutation of the SFTPC gene. The patient had fibrosing interstitial pneumonia, with HRCT pattern suggestive of nonspecific interstitial pneumonia, and unclassifiable pathology. Emphysematous lesions were remarkably apparent on imaging and pathologically in the vicinity of peribronchiolar fibrosis. The clinical presentation of the infant was comparable to that described previously.1 2 Interestingly, multiple lung cysts associated with septal thickening and ground glass opacities have been reported in subjects with familial pulmonary fibrosis carrying SFTPC mutations.3 4
The putative pathophysiology of SP-C-associated disease involves the dysfunction of surfactant homeostasis, causing injury or death of alveolar epithelial type II cells and myofibroblast proliferation. A process of genetically mediated alveolar injury may conceivably contribute to emphysema in addition to inflammation and fibrosis, and thus to the CPFE phenotype. As an auto-antibody was detected, a forme fruste of connective tissue disease may also have contributed to the lung disease.5
Adults with CPFE syndrome may have an underlying genetic predisposition. This hypothesis needs confirmation in further studies.
Supplementary materials
Web Only Data thx.2010.151407
Files in this Data Supplement:
Footnotes
Funding Hospices Civils de Lyon.
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.