Article Text

Abstract
Recent research suggests that mesenchymal stem cells (MSCs) are able to migrate specifically to tumours and their metastases throughout the body. This has led to considerable excitement about the possibility of modifying these cells to express anticancer molecules and using them as specific targeted anticancer agents. However, there are concerns that systemically delivered MSCs may have non-desirable effects, and there are also many unanswered questions including the mechanism of tumour homing. This review investigates the different MSC-delivered anticancer agents, addresses the questions and concerns, and tries to place this potential therapy in future cancer management.
- Mesenchymal stem cell
- lung cancer
- breast cancer
- TRAIL
- apoptosis
Statistics from Altmetric.com
Worldwide, cancer remains one of the leading causes of mortality and morbidity.1 The mainstay of cancer therapy includes treatment with surgery, chemotherapy and radiotherapy; however, despite improvements in treatment, many tumours remain unresponsive to traditional therapy. One of the many challenges of cancer treatment relates to the delivery of the antitumour therapy to the tumour site. It has recently been shown that bone marrow-derived stem cells (BMSCs) are able to migrate specifically to and incorporate within tumours, and this property can be used to deliver targeted anticancer therapies.
Bone marrow-derived stem cells
Stem cells have unlimited self-renewal properties with the ability to produce more differentiated progenitors. They include both embryonic and adult stem cells, of which BMSCs are the best characterised and most accessible. The BMSCs can be divided into the haematopoietic stem cells (HSCs), which produce progenitors for all types of mature blood cells, and mesenchymal stem cells (MSCs) which have an important role in supporting haematopoiesis and supplying differentiated stromal tissue.2
Contribution of BMSCs to tumour stroma
In addition to their involvement in haematopoiesis, BMSCs have also been shown to migrate to and participate in areas of new stroma formation throughout the body. This has been demonstrated particularly in areas of damage and repair, including simple wound healing, the fibrotic lung response to bleomycin and in the stromal tissue of tumours. In fact, tumours have been compared to unresolved wounds that produce a continuous source of inflammatory mediators.3 Bone marrow transplant experiments have provided much of the evidence for the participation of BMSCs in tumour tissue, with up to 40% of the myofibroblasts bone marrow-derived in murine studies4 5 and up to 20% of the neoplastic cells bone marrow-derived in a patient with lung cancer after sex-mismatched bone marrow transplantation.6
Exogenously delivered MSCs also localise preferentially to tumours, and this subgroup of cells is most likely to provide scope for future clinical translation as a cellular therapy without the necessity of a bone marrow transplant. We and others have shown that these cells preferentially migrate to tumour cells in vitro using transwell migration assays,7–10 and in vivo using animal tumour models. In these models, intravenously delivered MSCs have been shown preferentially to migrate to and survive in cancer tissue in breast and melanoma lung metastases,7 10–12 Kaposi's sarcoma (KS),13 colorectal cancer9 and gliomas.8 Other delivery routes have also demonstrated consistent MSC migration and tumour incorporation, including intraperitoneal delivery for ovarian cancer14 and intracerebrally in a glioma model.8
Mesenchymal stem cells
The MSC subgroup of cells is particularly suited to a role as a vector for cancer therapy. In contrast to BMSCs, they can be given as cellular therapy without the morbidity and mortality associated with bone marrow transplantation. This is because MSCs express the major histocompatibility complex (MHC) 1 but lack MHC 2 and the co-stimulatory molecules CD80, CD86, CD40,15 hence non-allogenic and xenograft MSCs could theoretically be used in immunocompetent patients. This has significant clinical implications, whereby engineered MSCs could be used in patients as a cell therapy without the considerations and complications surrounding immunomodulation associated with their use. This property could theoretically allow for the development of an MSC bank where allogenic cells could be stored and used for patients. Such standardised preparations of MSCs are being used in many clinical trials including cardiac disease, graft versus host disease (GvHD) and Crohn's disease, and the low immunogenicity has obviated the need for human leucocyte antigen (HLA) matching.16
In addition to the tumour-homing properties, MSCs are easily extracted and readily expandable, with up to 50 population doublings in 10 weeks.17 They are also easily transduced with integrating vectors due to their high levels of amphotropic receptors18 and provide long-term gene expression without affecting their MSC phenotype.19–21 There are no specific cell markers that can be used to categorically define an MSC, and present identification relies upon a combination of their ability to differentiate in vitro into fat, bone and cartilage, the expression of CD73, CD90 and CD105, and the lack of expression of haematopoietic cell markers (CD11b, CD14, CD19, CD34, CD45, CD79α, HLA-DR) on the cell surface.22
Homing mediators
The precise mechanism behind the specific homing of MSCs to the multiple tumours is difficult to pinpoint. The most likely cause of preferential migration is the release of chemotactic gradients from the tumours (figure 1). MSCs have a large variety of chemokine and cytokine receptors on their cell surface and respond functionally to the ligands in vitro. Furthermore, manipulation of these receptors and ligands has been shown to alter the migration patterns in vivo.23–27 Tumours are also known to produce a large array of chemokines and cytokines which could serve as ligands for the MSC receptors.28 Nevertheless, although migration mechanisms have been well characterised for haemopoietic stem cells with the ligand CXCL12 (stromal derived factor 1α, SDF1α) and its receptor CXCR4, the contributions of individual chemokines and cytokines in MSC migration have not been determined. Studies that have attempted to determine the mechanism of MSC migration have focused on investigating the chemokine receptors present on the cell surface of MSCs and their functional importance in vitro. Such an approach, however, has led to a lack of consensus between studies, with different receptors postulated (table 1).23–27

Mesenchymal stem cells (MSCs) home to tumours. MSCs are believed to extravasate similarly to leucocytes with adhesion molecules and integrins. They express very late antigen 4 (VLA-4) which binds to its counterpart adhesion molecule vascular cell adhesion molecule-1 (VCAM-1) on the endothelium. The expression of P-selectin has also been shown to be important for this process. MSCs migrate towards tumours in response to chemokines, which bind to the chemokine receptors on MSCs. The specific DiI-labelled (red) MSC homing is demonstrated in histological sections of lung metastases with (i) H&E and (ii) fluorescent microscopy of contiguous sections with DAPI (blue) nuclear counterstaining. Scale bars represent 20 μm.
Chemokine receptors expressed by mesenchymal stem cells (MSCs)
Of the chemokines, the contribution of CXCL12 to MSC recruitment is of particular interest. This chemokine is critical for haematopoietic stem cell migration and knockouts of either the ligand or receptor CXCR4 are universally fatal in utero, with the chemokine involved in the migration of HSCs from the liver to the bone marrow. Similarly, the migration of BMSCs to fibrotic lung is reduced with CXCL12 neutralisation.29 30 The importance of this chemokine in MSC migration is suggested by studies demonstrating an increased migration of MSCs, tranduced to overexpress CXCR4, to the infarcted myocardium,31 and the in vivo migration of MSCs to the brain after exogenous CXCL12 injection.32 CXCL12 may also be an important mediator of MSC recruitment to tumours. Breast cancer stromal tissue33 and whole tumour explants28 were shown to secrete this chemokine. Other studies, however, have failed to confirm the importance of CXCR4 for MSC migration. Blockade of CXCR4 did not affect MSC migration in a myocardial infarct model,34 and some groups have not been able to show expression of this receptor on MSCs.27
Based on the established literature for leucocyte migration, other possible important candidates for MSC chemotaxis include the receptors CXCR1,2 and CCR3, neutralisation of which has been shown to decrease leucocyte migration,35 36 and CCR2, important in macrophage trafficking.37 Indeed, CCL2 (monocyte chemoattractant protein 1, MCP-1) was shown to be secreted by breast cancer cells in vivo and the use of a neutralising antibody reduced the homing of MSCs.28 The same study also demonstrated an increased serum level of CCL2 in post-menopausal patients with breast cancer compared with controls.
MSCs are also believed to use similar mechanisms to leucocytes for extravasation from the vasculature with the use of adhesion molecules and integrins (figure 1).38 P-selectin is important in the initial endothelial contact and rolling of leucocytes, and neutralising antibodies against the endothelially expressed P-selectin also resulted in fewer endothelial cell-bound MSCs in vitro.38 The very late antigen-4 (VLA-4) and its counterpart adhesion molecule vascular cell adhesion molecule-1 (VCAM-1) are critical for leucocyte arrest on activated endothelium, and antibodies to either resulted in decreased MSC adherence to the endothelium.38 Furthermore, the migration of MSCs to ischaemic myocardium was reduced by blocking MSC β1-integrin, a component of VLA-4.34 Despite the similarities, there are however likely to be differences between leucocyte and MSC extravasation—for example, platelet/endothelial cell adhesion molecule-1 (PECAM-1), which is involved in leucocyte transmigration, is not expressed on MSCs.39
The variability between the studies of the chemokines and cytokines relevant to MSC migration may be partly explained by the lack of homogeneity of the MSC population. This includes the extraction and identification of the cells, the culture conditions and the possibility of cell activation either in vivo or ex vivo. In vitro passaging has been shown to reduce chemokine receptor expression and MSC migratory potential.25 40 Culture confluence has also been shown to impact on MSC homing, with increased confluency increasing the production of the tissue inhibitor of matrix metalloproteinase-3 (TIMP-3) and inhibiting transendothelial migration.41 Conversely, hypoxic culture conditions increase matrix metalloproteinases (MMPs) and MSC migration.42 Despite the variability, the general consensus is that MSCs do express a number of chemokine receptors which are likely to be involved in their homing capabilities,43 often with a combination of cytokines and chemokines necessary for the maximal effect.44
MSCs as delivery vectors for cancer therapy
The ability of MSCs to specifically migrate to tumours has led several groups to investigate these cells in delivering anticancer agents (table 2). Human MSCs have been engineered to express and provide targeted delivery of interferon β (IFNβ) to gliomas8 and metastatic breast,12 melanoma11 12 and prostate46 cancer models. The use of MSC-delivered IFNβ, which suppresses tumour cell growth by induction of cancer cell differentiation, S-phase accumulation and apoptosis, resulted in increased survival and/or reduced tumour burden in all these models. IFNα-expressing MSCs have also been used for the immunostimulatory, apoptosis-inducing and anti-angiogenic effects of IFNα with similar results in a metastatic melanoma model.45 MSCs have also been adenovirus-transduced to express the immunostimulatory interleukin (IL)12 to improve immune surveillance against cancer cells. In this model, the intravenously delivered IL12-expressing MSCs reduced metastases from subcutaneous tumours.49 In addition, if delivered intraperitoneally 1 week before tumour inoculation, the MSCs were able to prevent subcutaneous tumour development.50 A similar approach of immunostimulation with tumour-targeted chemokines was used to deliver the chemokine CX3CL1 (fractalkine) which is able to activate T cells and NK cells. This therapy led to a reduction in the number of lung metastases following intravenous delivery of colon cancer and melanoma cells.10 MSCs have also been produced to deliver IFNγ which stimulated apoptosis and inhibited leukaemic cell proliferation in vitro,47 and the immunomodulatory cytokine IL2 with a reduction in tumour growth and improved survival when directly injected into murine gliomas.48
Anticancer agents delivered by mesenchymal stem cells (MSCs)
In addition to the delivery of growth factors, cytokines and chemokines, MSCs have also been engineered to deliver conditional replicative adenoviruses to reduce tumour growth and spread in vivo. These viruses are able to destroy tumour cells by viral replication and, following oncolysis, further newly produced virus is then released to the surrounding tumour tissue. This therapy improved survival in a murine ovarian cancer model,14 orthotopic breast and lung tumour models,55 and a lung metastasis model.56
MSCs have also been used to serve as vehicles for targeted chemotherapy with an enzyme prodrug conversion approach. One study combined MSCs that produced herpes simplex virus-thymidine kinase (HSV-tk) retroviral vectors in the proximity of tumours with the prodrug ganciclovir, leading to glioma cell death in vitro and in vivo.51 In different studies, MSCs expressed the cytosine deaminase enzyme which converts the substrate 5-fluorocytosine (5-FC) to the highly toxic 5-fluorouracil (5-FU); delivery to the tumour cells resulted in a reduction of melanoma52 and colon cancer53 subcutaneous tumours. However, these approaches are limited by the toxicity of the treatment to the delivery MSC.
We have used a conditional lentivirus to transduce MSCs to express TRAIL under the control of doxycyline.7 TRAIL is a type 2 transmembrane protein with homology to other members of the tumour necrosis factor (TNF) family.59 60 This novel ligand was found to cause apoptosis selectively in tumour cells, sparing normal cells. The mechanism underlying this selectivity is not fully understood, but it is likely to be multifactorial and to include the presence of active and decoy TRAIL receptors, post-translational receptor modification and the activation of non-apoptotic signalling pathways (figure 2). Delivery of this agent has advantages over some of the other anticancer MSC delivery systems described above as there appear to be very limited effects on non-cancerous tissues. Indeed, TRAIL therapy in the form of recombinant protein and monoclonal antibodies to DR4 or DR5 has been used in phase 2 clinical trials with good initial safety and tolerability data.61–64 The targeted delivery of TRAIL with MSCs overcomes some of the problems encountered with recombinant TRAIL or TRAIL monoclonal antibodies. MSC-targeted TRAIL is more physiological than the monoclonal antibodies, which rely on the effects of a specific active receptor, and has a much better half-life and increased delivery across the blood-brain barrier than recombinant TRAIL. In addition, the MSCs deliver TRAIL directly to the tumours. In our murine models, cellular therapy with TRAIL-expressing MSCs led to a reduction in subcutaneous tumour growth and reduced, and in a significant number of mice eliminated, the development of lung metastases.7
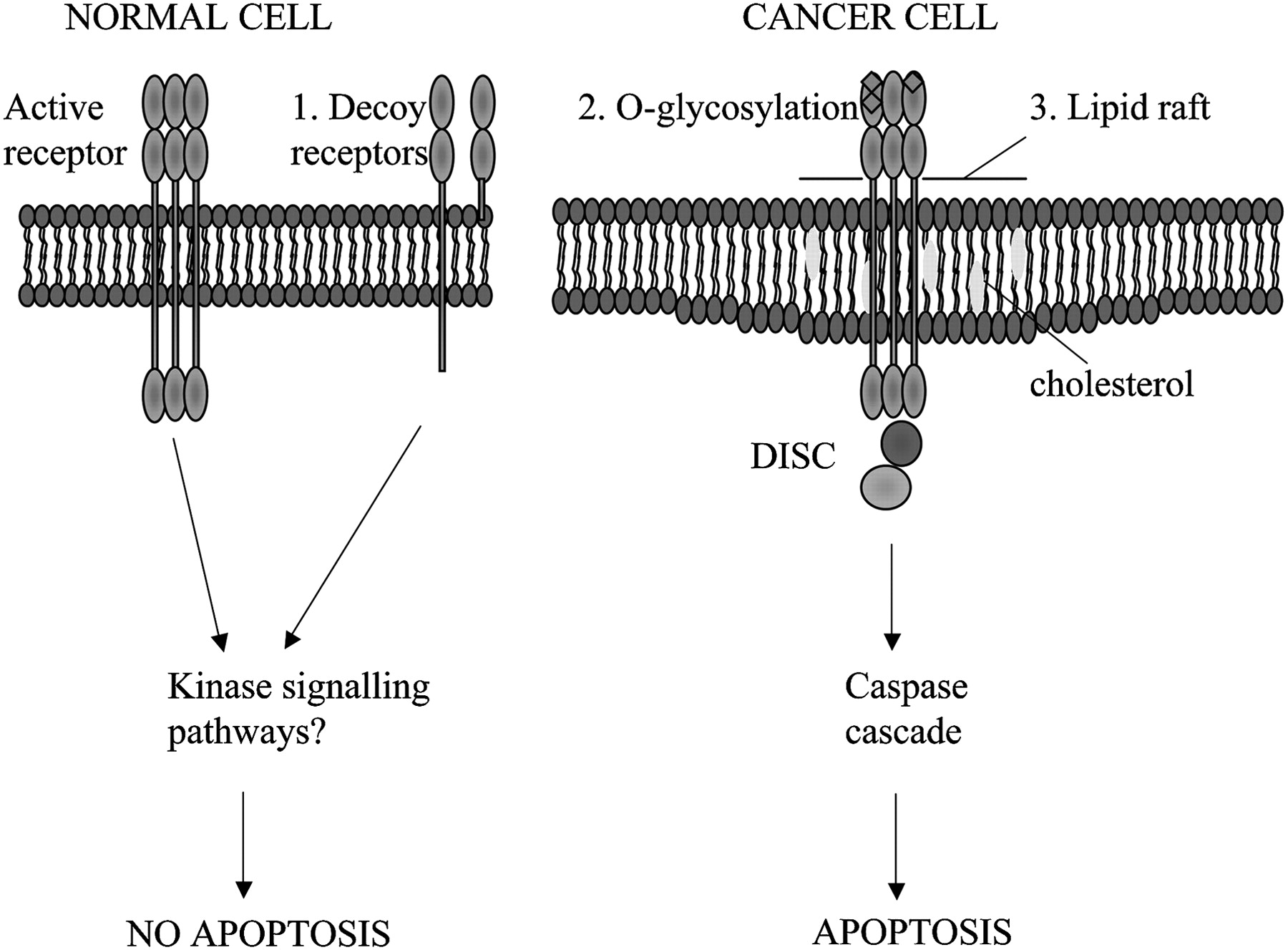
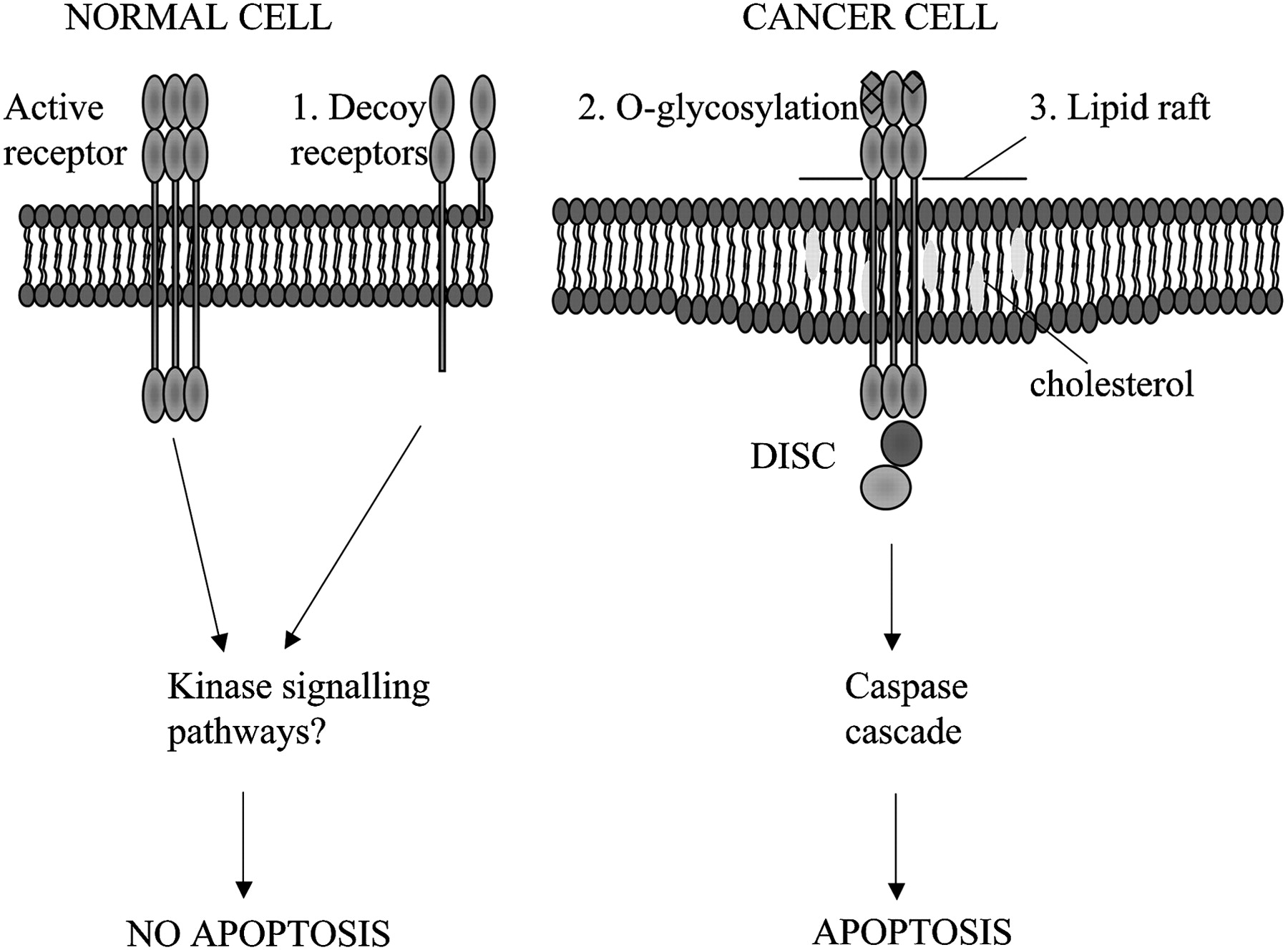
Cancer cells are specifically sensitive to TRAIL-induced apoptosis. Possible mechanisms to explain the selective apoptosis of cancer cell to TRAIL include (1) increase in decoy receptors (which lack the intracellular death domain) in normal cells which may activate anti-apoptotic kinase signalling pathways, (2) O-glycosylation of receptors and (3) their location in lipid rafts enhancing death inducing signalling complex (DISC) formation.
MSCs—more than an inert drug vehicle: advantages
The ability of MSCs to specifically localise to multiple tumours makes them extremely attractive for directed cancer therapy. However, MSCs are not simple inert vectors but active cells with effects on both physiological and pathological processes. In particular, it is known that MSCs have profound immunosuppressive effects. T cells cultured with MSCs do not proliferate with antigenic or mitogenic stimuli. This may be due to the arrest of the T cell in the G0/G1 phase with consequent prevention of S phase entry and inhibition of cell division.65 Similar effects have been demonstrated on B cells66 and dendritic cells,67 leading to a reduction in plasma cell maturation and antibody production and antigen presentation, respectively. In addition to direct T cell inhibition, MSCs also induce CD4+CD25+FoxP3+ regulatory T cells (Tregs) which further limit the activation of CD4 and CD8 subsets, B cells and NK cells.68 MSCs affect immune cells by several mechanisms including direct cell contact and the release of many soluble factors, including nitric oxide.69 These immunosuppressive effects of MSCs have been harnessed in the treatment of GvHD following bone marrow transplantation.70
In addition, MSCs are thought to be able to reduce damage in several injury systems by both stimulating repair and exerting anti-inflammatory effects. This has been used in clinical trials in cardiology71–74 whereby MSCs have been infused acutely following myocardial infarction and in chronic ischaemic heart failure, with an improvement in cardiac function, infarct size and remodelling.74 The paracrine effects of MSCs on tissue protection and repair, with the secretion of trophic factors, have also been proposed for the improvements in the lung following bleomycin-induced pulmonary fibrosis in murine models.75 76 Expression of IL1 receptor antagonist by MSCs and the subsequent reduction of the proinflammatory cytokines TNFα and IL1 have been postulated as central to this effect. Phase 2 clinical trials are presently being performed to investigate whether the anti-inflammatory and reparative function of MSCs may benefit patients with moderate to severe chronic obstructive lung disease.
MSCs also appear to have intrinsic antitumour properties (figure 3). They have been shown to arrest hepatoma, lymphoma and insulinoma cells at G0/G1, and this reduced proliferation was accompanied by an increase in cancer cell apoptosis and a reduction in malignant ascites when hepatoma cells were injected intraperitoneally.77 An improvement in other cancer models has also been demonstrated with the use of MSCs. Intratumoural injection of MSCs led to the inhibition of tumour growth and increased survival of rats with glioma,48 and both intratumoural and intravenous injections of MSCs led to the reduction in growth and metastases of a breast cancer model with the increase of apoptotic markers.78

Mesenchymal stem cells (MSCs) may have both growth inhibitory and stimulatory effects on tumours. The literature is divided on the impact that MSCs have on tumour growth with both (A) growth stimulatory and (B) inhibitory effects described. (A) Possible mechanisms for increased tumour proliferation include (i) reduced immunosurveillance secondary to MSC-induced T cell, B cell and natural killer (NK) cell suppression; (ii) promotion of motility, invasion and metastasis of cancer cells by CCL5 release; and (iii) interleukin 6 (IL6) release leading to STAT-3 phosphorylation and tumour cell proliferation. (B) Possible mechanisms for tumour inhibition include (i) promotion of cancer cell cycle arrest and apoptosis; (ii) inhibition of Akt activity; (iii) downregulation of nuclear factor κB (NFκB); and (iv) downregulation of the Wnt pathway by the release of the soluble inhibitor Dickkopf1 (DKK1), both of which reduce cancer cell proliferation.
Furthermore, intravenously delivered MSCs were able to inhibit the growth of KS in a mouse model due to cell–cell contact-induced inhibition of Akt activity within KS cells.13 The release of soluble factors by MSCs has also been shown to reduce tumour growth and progression in glioma,48 melanoma and lung carcinoma models,79 and conditioned media from MSCs have been shown to lead to the downregulation of NFκB in hepatoma and breast cancer cells resulting in a decrease in their in vitro proliferation.80 This group delineated a further mechanism with the secretion of dickkopf proteins (DKK-1) by MSCs leading to the downregulation of the Wnt signalling pathway in breast cancer cells and a reduction in their proliferation.81
MSCs—more than an inert drug vehicle: disadvantages
It is, however, especially important to consider the role of MSCs in specific cancer types in the context of their immunosuppressive, reparative and angiogenic properties (figure 3). In contrast to the antitumour effects discussed above, subcutaneously-delivered allogenic melanoma cells only produced tumours in mice with the co-administration of MSCs, and immunosuppression was thought to be a pivotal factor for this observation.82 The same group also demonstrated an earlier development of tumours when syngeneic Renca kidney cancer cells were implanted with MSCs. However, MSCs did not alter cancer cell growth in vitro, supporting an immunosuppressive role for the in vivo differences. The kinetics of tumour growth and metastases were not affected in these experiments.83
The production of trophic factors by MSCs has also been implicated in enhancing tumour growth and spread. MSCs enhanced the in vivo growth of Burkitt's lymphoma cells by a mechanism dependent on the VEGF pathway.84 MSCs have also been shown to secrete the chemokine CCL5 which promoted metastasis of breast cancer cells in a mouse subcutaneous xenograft model.85 The production of IL6 by MSCs was also implicated in the increased growth of breast cancer cells by STAT-3 phosphorylation and signalling.86 Further tumour-enhancing effects of MSCs have also been demonstrated with colonic cancer87 and chronic myeloid leukaemia cell lines. In the latter experiment, the MSCs increased in vivo tumour proliferation but reduced cancer cell proliferation in vitro. In order to resolve the discrepancy, the authors suggested that MSCs were able to downregulate cyclin D2 and arrest cancer cells at G0/G1, preserving their proliferative capacity and reducing apoptosis.88
A further concern of the use of MSCs as an anticancer delivery agent is the potential of these cells for malignant change, particularly in view of their unlimited capacity for proliferation. Karyotype abnormalities have been demonstrated with in vitro passage,89–91 and malignant change of bone marrow-derived cells has also been implicated in a murine gastric carcinoma model.92 However, a recent study performed to assess the potential susceptibility of human bone marrow-derived MSCs to malignant transformation demonstrated no features consistent with this, with stable karyotypes and shortening teleomeres over the 44-week culture period, and concluded that MSCs remained suitable for cell therapies after in vitro expansion.93
Clinical translation
The ultimate goal of this area of research is for the development of a cellular therapy for humans. MSC-directed anticancer treatment has definite potential for translation to clinical medicine. The specific targeting is likely to produce significantly less host toxicity than traditional agents and will allow much higher local concentrations of antitumour agents. Other tumour-directed therapies such as monoclonal antibodies rely on the detection and expression of specific tumour antigens which are likely to change between patients, cancers and time. Conversely, MSCs appear to be able to localise specifically in a range of different tumours and their metastases.
The most likely position for MSC-delivered therapy in cancer treatment would be in combination with present radiotherapy and chemotherapy agents. Our study demonstrated an elimination of metastases with TRAIL-expressing MSCs,7 and others have also described a reduced metastatic load with MSC-delivered therapies (table 2). This effect on metastases is extremely important as secondary spread is the main cause of mortality and morbidity in patients with cancer. Many patients with solid organ tumours progress to metastatic disease despite primary tumour resection, chemotherapy and radiotherapy. The recurrence rate following surgery of curative intent, despite tumour negative lymph nodes, is up to 40% in non-small cell lung cancer, 30% in colon cancer, 25% in breast cancer and 15–50% in prostate cancer.94 The use of new molecular and cytological techniques to detect small numbers of circulating malignant cells makes the identification of patients at significant risk of future metastatic disease a possibility.94 95 The detection of these cells, presumably resistant to the host's endogenous immune surveillance, is related to metastatic recurrence and poorer prognosis and could be amenable to MSC-directed therapy.
There may also be an increased synergism of a combination therapy approach which is greater than the sum of its parts (figure 4). The homing of MSCs to tumours has been shown to increase significantly with the use of radiation. This has been demonstrated with irradiated glioma, breast and colon cancer xenograft models, in addition to a syngeneic murine breast carcinoma model, and to be secondary to the increased inflammation and expression of cytokines from the irradiated tissue in addition to the upregulation of chemokine receptors on the MSCs.96 97 Chemotherapeutic agents and radiotherapy have also been shown to increase the cancer-killing effects of some of the MSC-delivered therapies. The apoptotic effects of TRAIL are significantly increased in vitro and in in vivo xenograft studies with chemotherapy or radiotherapy. A number of possible mechanisms have been postulated for this synergism, including the upregulation of TRAIL receptors,98 99 the clustering of TRAIL receptors into lipid rafts,100 the downregulation of apoptotic pathway inhibitors101 or the enhanced cleavage of caspases.102 103

{kind=link}
{kind=link}
{kind=link}
{kind=link}
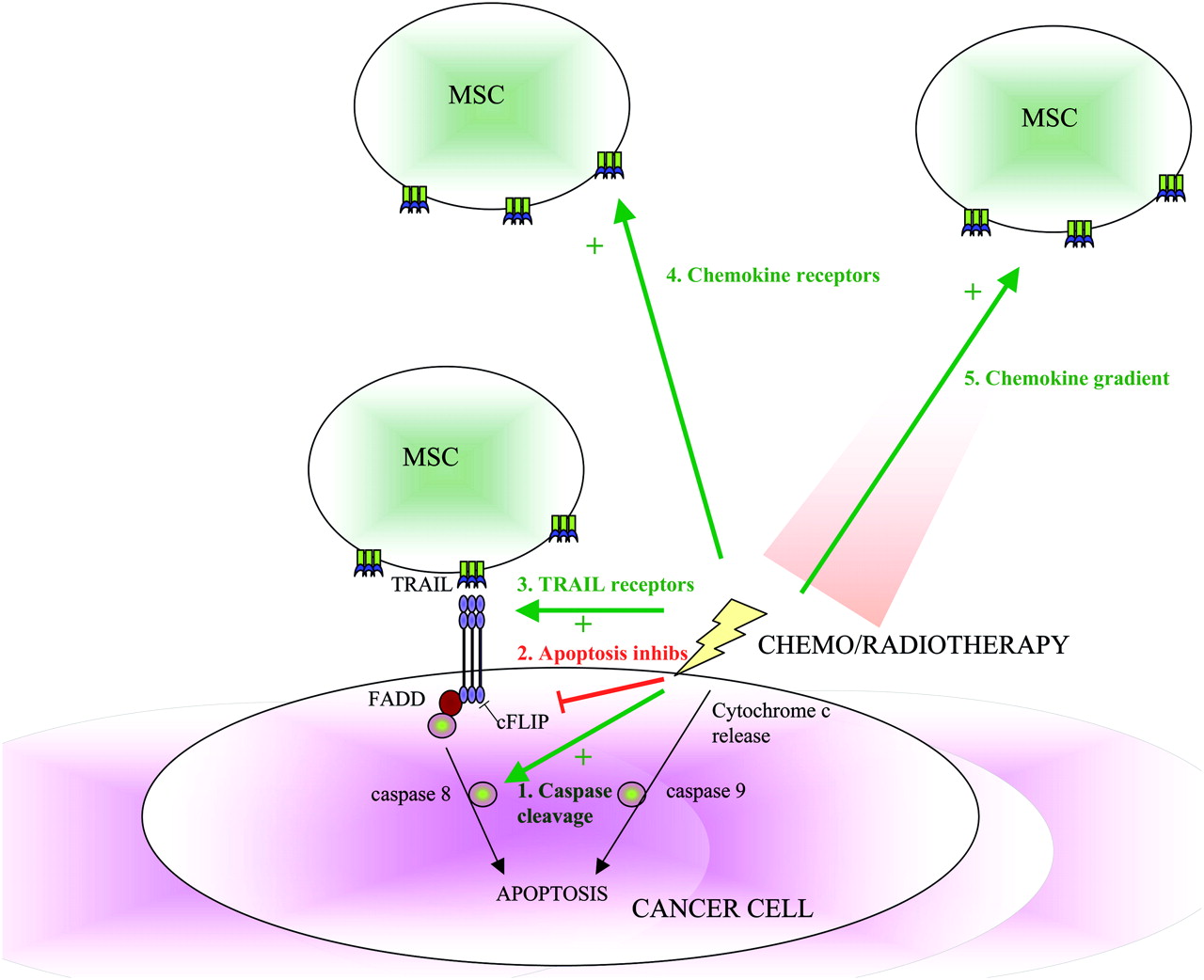
Synergy of chemotherapy and radiotherapy with mesenchymal stem cell (MSC)-delivered anticancer agents. Chemotherapy and radiotherapy cause DNA damage and cancer cell apoptosis by the intrinsic apoptosis pathway, whereas TRAIL leads to cancer cell apoptosis by the extrinsic apoptosis pathway. Chemotherapy and radiotherapy can enhance the TRAIL sensitivity of cancer cells by (1) activating caspase cleavage; (2) inhibiting apoptotic inhibitors (eg, c-Flice-like inhibitory protein (c-FLIP)); and (3) increasing TRAIL receptor number and their location within the lipid raft. Radiotherapy can increase the homing of systemically-delivered MSCs to tumours by increasing (4) MSC chemokine receptor expression and (5) chemokine release by the tumours.
As described above, there are some concerns for MSC-based therapies. Despite these, however, phase 1 and 2 clinical trials have now been performed or are ongoing, with exogenous MSCs for the promotion of haematopoietic recovery,104 for GvHD following bone marrow transplantation,70 osteogenesis imperfecta,105 ischaemic cardiac disease,74 Crohn's disease16 and chronic obstructive pulmonary disease,106 and so far neither acute nor long-term adverse effects have been reported following their infusion.
Conclusions
MSCs are well placed to be used as vectors for anticancer treatment. They are easily accessible and tranducible, and have been shown to migrate to tumours in a variety of studies and models. Their theoretical potential for malignant transformation and ability to increase cancer growth and metastases in some models has correctly led to significant caution. It is encouraging, however, that recent studies have downplayed the malignant potential of these cells.93 It is also important to note that, although it is unclear from the present research reports whether MSCs have a positive or negative effect on tumour growth when used as an isolated therapy, the literature is much more consistent with respect to the documented antitumour effects of MSCs used as vectors to deliver specific anticancer agents (table 2). These studies have documented positive effects in vivo, including the reduction in tumour growth, elimination of metastases and improvement in survival. The success of this area of work has produced excitement that this therapy may have clinical application and, although there are many unanswered basic science questions remaining, including the optimal timing of delivery, the number of cells needed, and the mechanism of homing, this has not prevented the development of clinical trials with MSCs in other areas with initial encouraging results and a lack of adverse effects. Finally, it is also clear that new modalities of cancer treatments are urgently needed for a desperate disease which may push this research to the clinic.
References
Footnotes
Funding MRL is a Medical Research Council (MRC) UK Clinical Training Fellow. SMJ was an MRC Clinician Scientist. This work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.