Article Text

Abstract
Background Mechanical ventilation (MV) is a life-saving intervention in acute respiratory failure without any alternative. However, even protective ventilation strategies applying minimal mechanical stress may evoke ventilator-induced lung injury (VILI). Adjuvant pharmacological strategies in addition to lung-protective ventilation to attenuate VILI are lacking. Adrenomedullin exhibited endothelial barrier-stabilising properties in vitro and in vivo.
Methods In untreated mice (female C57/Bl6 mice, 11–15 weeks old) and animals treated with adrenomedullin, lung permeability, local and systemic inflammation and markers of distal organ function were assessed following 2 or 6 h of mechanical ventilation with 100% oxygen and protective or moderately injurious ventilator settings, respectively.
Results Adrenomedullin dramatically reduced lung permeability in VILI in mice, leading to improved oxygenation. Adrenomedullin treatment reduced myosin light chain phosphorylation, attenuated the accumulation of leucocytes in the lung and prevented the increase in lactate and creatinine levels in mice ventilated with high tidal volumes. Moreover, adrenomedullin protected against VILI even when treatment was initiated 2 h after the beginning of mechanical ventilation in a 6 h VILI mouse model.
Conclusion Adjuvant treatment with adrenomedullin may be a promising new pharmacological approach to attenuate VILI.
- Ventilator-induced lung injury
- adrenomedullin
- acute lung injury
- permeability
- myosin light chain
- renal failure
- microcirculation
- ARDS
- pulmonary oedema
Statistics from Altmetric.com
- Ventilator-induced lung injury
- adrenomedullin
- acute lung injury
- permeability
- myosin light chain
- renal failure
- microcirculation
- ARDS
- pulmonary oedema
Introduction
In acute respiratory failure, mechanical ventilation (MV) is a life-saving treatment without alternatives, and MV is also employed following surgery or trauma. One-third of all patients in intensive care units worldwide receive MV.1 However, even minimal MV-induced physical forces on lung tissue may evoke ventilator-induced lung injury (VILI), an important undesirable effect of respirator treatment.2 VILI is characterised by pulmonary inflammation with liberation of cytokines, recruitment of leucocytes to the lung and increased lung permeability, resulting in lung oedema, surfactant dysfunction, impaired lung compliance and deterioration of pulmonary gas exchange.3 A clinical study of the Acute Respiratory Distress Syndrome (ARDS) network revealed that minimisation of MV-induced physical stress by reduction of tidal volumes to 6 ml/kg significantly improved clinical outcome of mechanically ventilated patients.4 However, preinjured lungs in particular are sensitive for the development of VILI even in the setting of lung-protective ventilation.5 Moreover, the need for hyperoxic ventilation of preinjured lungs may further aggravate lung inflammation and barrier dysfunction.6 As the necessity to guarantee sufficient gas exchange frequently limits a further substantial reduction of tidal volumes and oxygen supply, new adjuvant pharmacological treatments in addition to lung-protective ventilation are needed to prevent VILI.
Adrenomedullin (AM), an endogenous 52 amino acid peptide belonging to the calcitonin gene-related peptide family, plays a crucial role in endothelial barrier integrity, which is underlined by the observation that AM deficiency in mice is lethal due to midgestation death as a result of hydrops fetalis.7 AM acts via binding of the calcitonin receptor-like receptor, thereby raising intracellular cAMP levels in endothelial cells and reducing myosin light chain (MLC) phosphorylation. Thus, AM may prevent endothelial contraction and intercellular gap formation, which are ultrastructural correlates of endothelial permeability.8–10 Notably, AM expression is upregulated in inflammatory diseases including acute lung injury (ALI) and sepsis, and endogenous AM may contribute to the protection of vascular function in inflammation.8 Moreover, exogenous AM reduced pulmonary hyperpermeability, improving pulmonary gas exchange in experimental models of ALI and sepsis.9 11–13 Furthermore, AM treatment limited hyperinflammation in experimental sepsis and protected against microcirculatory impairment.14 15
Pulmonary hyperpermeability, compromised microcirculation, and pulmonary and systemic inflammation crucially contribute to the pathophysiology of VILI and VILI-associated multiorgan dysfunction,3 16 and exogenous AM may possibly be beneficial during MV. We therefore hypothesised that AM may prevent VILI, particularly due to protection against VILI-associated endothelial barrier dysfunction.
VILI is likely to impact the outcome of mechanically ventilated patients with ALI/ARDS. Thus, future strategies for the treatment of ALI/ARDS should also consider VILI. Recent work of our group and other groups identified AM as a promising candidate for an adjuvant pharmacological approach to treat ALI/ARDS. Notably, its potential translation into clinical trials is supported by the recent positive opinion of the Committee for Orphan Medicinal Products of the European Medicines Agency (EMA), which recommends the granting of the development of AM as an orphan medicinal product for the treatment of ALI (EMA/COMP/104704/2010 to SH of our group). Therefore, the results of the current study may impact on clinical trial design in the near future.
Materials and methods
Mechanical ventilation
All procedures were approved by local authorities. Female C57/B6 mice (11–15 weeks; Charles River, Sulzfeld, Germany) were used for this study. In the 2 h experiments mice were anaesthetised by repetitive intraperitoneal injections every 50 min of xylazine (0.025 mg/g) and ketamine (0.125 mg/g). Mice ventilated for 6 h were anaesthetised by intraperitoneal injections of fentanyl (0.075 mg/kg), midazolam (1.5 mg/kg) and medetomedin (0.75 mg/kg). Repetitive applications of fentanyl (0.016 mg/kg), midazolam (0.33 mg/kg) and medetomedin (0.16 mg/kg) were done via an intraperitoneal catheter when required. In all groups body temperature was measured continuously using a rectal probe and maintained at 37°C by a heating pad. After tracheotomy and intubation, mice were ventilated (MiniVent, Harvard Apparatus, March, Germany), and airway pressure was raised to 35 cm H2O for 5 s for lung recruitment, followed by ventilation with 7 ml/kg tidal volume, positive end expiratory pressure (PEEP)=6 cm H2O and fractional inspired oxygen (Fio2)=1 (MiniVent, Hugo Sachs Elektronik, March, Germany). A carotid artery catheter was placed for blood pressure monitoring17 and continuous infusion of 0.9% NaCl containing 100 mmol/l HCO3– at 350 μl/h throughout the ventilation period.
When baseline conditions were established after preparation, ventilation in low tidal volume (LVT) groups was continued as described. In high tidal volume (HVT) groups, settings were switched to 17 ml/kg tidal volume and PEEP=0 cm H2O. In the groups ventilated for 6 h (6h vent) respirator settings were 12 ml/kg tidal volume, PEEP=2. Performing blood gas analysis in preliminary experiments, respiratory rates of 240, 95 and 120/min in LVT, HVT and 6h vent groups, respectively, were determined to maintain pH at 7.4±0.1. Peak airway pressure at the end of the ventilation protocol was 13.0±0.5 cm H2O in the LVT, 19.0±1.3 cm H2O in HVT and 18.6±0.6 cm H2O in the 6 h vent group. At termination of experiments, mice were sacrificed by exsanguination via the carotid catheter. As a non-ventilated control group, spontaneously breathing mice (Fio2=0.21), which were not tracheotomised and did not undergo arterial catheterisation, were employed.
AM treatment
AM (0.05 mg/kg/h; Phoenix Pharmaceuticals, Burlingame, California, USA) was continuously infused via the carotid catheter. In the 2 h experiments AM infusion was started with the onset of the ventilation protocol. In the 6 h experiments AM infusion was started 2 h after the onset of MV. In both protocols AM infusion was continued until the termination of the ventilation protocol. The chosen dose of AM was extrapolated from experiments of our group performed in rats previously.13
Lung vascular permeability
Human serum albumin (HSA; 1 mg; Baxter, Unterschleißheim, Germany) was injected via the carotid artery catheter or the tail vein in ventilated or non-ventilated mice, respectively, 90 min before termination of the experiment. Mice were sacrificed and bronchoalveolar lavage (BAL) was performed with 2×800 μl of phosphate-buffered saline (PBS). In 6h vent groups and corresponding controls, only the right lung was lavaged with 4×400 μl of PBS. In all groups, the BAL recovery rate was 91±4%. BAL and plasma HSA levels were quantified by ELISA (Bethyl Laboratories, Montgomery, Texas, USA). Permeability was assessed by calculating the HSA BAL/plasma ratio.
In separate experiments, 20 mg/kg Evans Blue dye (EBD) (Fluka, Schnelldorf, Germany) was injected 10 min before the end of the experiment. Mice were sacrificed, and lungs were flushed and analysed for EBD content by infrared imaging (Li-Cor Odyssey, Lincoln, Nebraska, USA).13
Measurement of oxygenation, lactate, creatinine, aspartate aminotransferase (AST) and alanine aminotransferase (ALT)
Arterial oxygen pressure (Pao2) and lactate were quantified in arterial blood of all animals subjected to MV (ABL700, Radiometer-Copenhagen, Denmark). Oxygenation was expressed as Pao2/Fio2 (P/F ratio). Creatinine, AST and ALT were quantified by routine laboratory test.
MLC phosphorylation
MLC phosphorylation was assessed by western blot. Snap-frozen lungs were homogenised, lysed in buffer containing Triton X-100, subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and blotted for 45 min. Membranes were exposed to rabbit phospho-specific MLC (Thr18/Ser19; Cell Signalling, Boston, Massachusetts, USA) and goat antiactin (Santa Cruz, Santa Cruz, California, USA) antibody, respectively, and subsequently incubated with secondary antibodies (Cy5.5-labelled antirabbit, IRDye 800-labelled antigoat) (Rockland, Gilbertsville, Pennsylvania, USA) as described.11 Proteins were detected simultaneously by infrared imaging (Li-COR Odyssey).
BAL and blood leucocyte counts
BAL cells were counted by haemocytometer and differentiated by flow cytometry (FACSCalibur, BD, Heidelberg, Germany) analysing forward and side scatter characteristics and expression of surface antigens CD45, Gr-1 and F4-80. Blood leucocytes were counted and differentiated by flow cytometry analysing CD45 and Gr-1 surface expression using TruCount tubes (BD, Heidelberg, Germany).
Cytokines in BAL and plasma
Interleukin 6 (IL-6), macrophage-inflammatory protein-1α (MIP-1α), MIP-2 and keratinocyte-derived chemokine (KC) levels in BAL and blood plasma were quantified by a multiplex assay as described previously according to the manufacturer's instructions (BioRad, Hercules, California, USA).18
Data analysis
Data are expressed as mean±SEM. For comparison between groups, one-way analysis of variance followed by Newman–Keuls post-test was used. p-Values<0.05 were considered significant.
Results
AM treatment attenuated VILI-associated lung vascular hyperpermeability
Both LVT and moderate HVT provoked a marked increase of lung vascular permeability after 2 h of MV (figure 1A–C). Systemic application of AM attenuated pulmonary hyperpermeability as shown by a significant decrease of HSA flux into the alveolar space (figure 1A), as well as EBD extravasation (figure1B,C).

Adrenomedullin (AM) treatment attenuated ventilator-induced lung injury (VILI)-associated lung vascular permeability. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT). Intervention groups were treated with AM (NV, non-ventilated mice). (A) Human serum albumin (HSA) was infused 90 min prior to termination of the experiment. HSA levels in plasma and bronchoalveolar lavage (BAL) were determined. An increased HSA BAL/plasma ratio indicated microvascular leakage (###p<0.001 vs NV; *p<0.05, **p<0.01, ***p<0.001; NV n=6, LVT n = 10, LVT+AM n=6, HVT/HVT+AM n=5). (B) Evans Blue dye (EBD) was infused 10 min prior to termination of the experiment. Lungs of ventilated mice displayed EBD extravasation indicating pulmonary microvascular leakage, which was prevented by AM treatment. (C) EBD content was determined by infrared imaging. (One representative image out of three in every group is shown.)
AM treatment improved oxygenation in VILI
HVT-associated pulmonary hyperpermeability was accompanied by a decrease of the P/F ratio within 2 h of ventilation as compared with the LVT group, and AM increased oxygenation under HVT from 317±31 mm Hg in untreated mice to 343±32 mm Hg in AM-treated mice (p<0.05) (figure 2).

Adrenomedullin (AM) treatment improved oxygenation in ventilator-induced lung injury (VILI). Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT). Intervention groups were treated with AM. HVT ventilation was accompanied by a marked decrease of the Pao2/Fio2 (P/F) ratio within 2 h of ventilation compared with the LVT group, and treatment with AM prevented oxygenation impairment (*p<0.05; LVT n=15, LVT+AM n=17, HVT n=12, HVT+AM n=12).
AM treatment prevented VILI-associated phosphorylation of the MLC in the lung
MLC phosphorylation-dependent endothelial cell contraction reportedly contributes to paracellular fluid flux and subsequent oedema formation.19 VILI-induced alteration of pulmonary permeability was associated with MLC phosphorylation in our model (figure 3). AM treatment reduced this in vivo MLC phosphorylation in both the LVT and HVT group, as demonstrated by western blot analysis (figure 3).
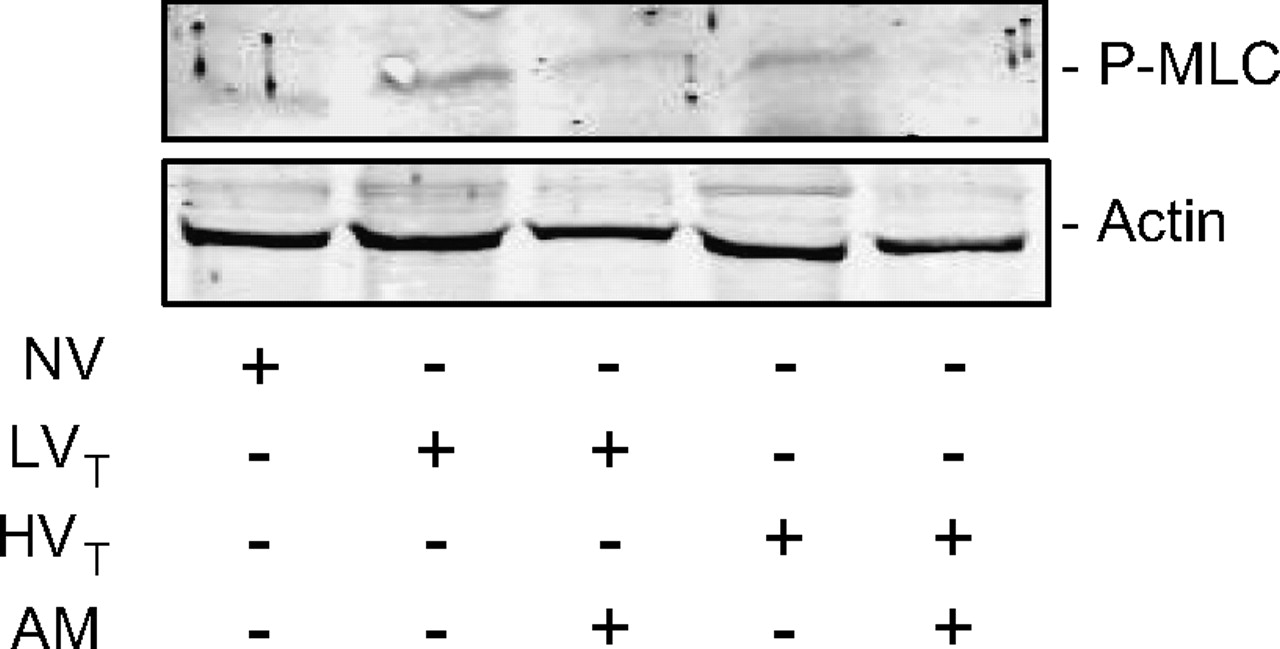
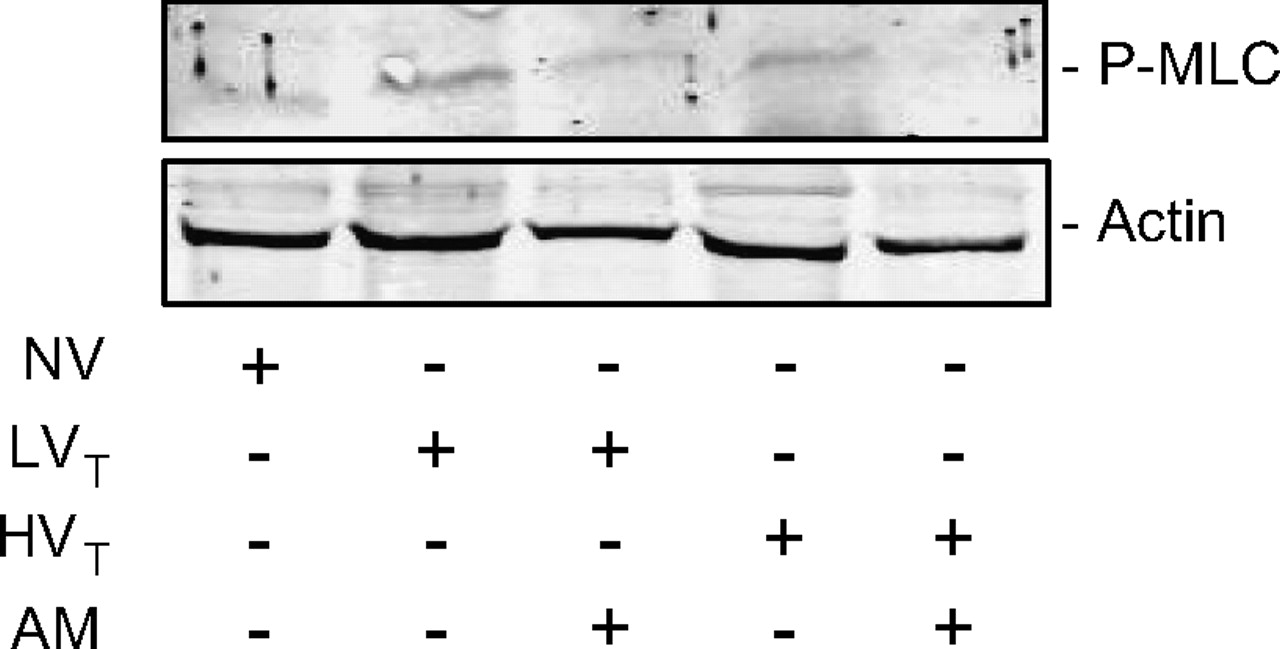
Adrenomedullin (AM) treatment prevented ventilator-induced lung injury (VILI)-associated phosphorylation of myosin light chain (MLC) in the lung. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT). Intervention groups were treated with AM (NV, non-ventilated mice). AM treatment reduced VILI-associated in vivo MLC phosphorylation in both the LVT and HVT group, as demonstrated by western blot analysis. (One representative experiment out of three in every group is shown.)
AM treatment decreased alveolar leucocyte counts
The numbers of leucocytes and particularly polymorphonuclear leucocytes (PMNs) in BAL fluid (BALF) were significantly increased in the untreated HVT group. AM treatment resulted in a substantial decline of pulmonary leucocyte and PMN numbers in BALF of HVT mice (figure 4A). To analyse further whether reduced BALF cell numbers were due to local or systemic AM action, blood cell counts were analysed. HVT elicited a significant rise in peripheral blood leucocyte and PMN counts. In the AM-treated group, significantly higher PMN counts as compared with the untreated HVT group were observed (figure 4B).

Adrenomedullin (AM) treatment decreased leucocyte influx into the lung. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT). Intervention groups were treated with AM (NV, non-ventilated mice). (A) AM reduced pulmonary leucocyte influx induced by HVT ventilation. Lungs were lavaged and leucocyte and polymorphonuclear leucocyte (PMN) counts in the bronchoalveolar lavage (BAL) were determined (##p<0.01, ###p<0.001 vs NV, *p<0.05, **p<0.01; NV n=6, LVT n=6, LVT+AM n=10, HVT/HVT+AM n=6). (B) Increased leucocyte and PMN cell counts in blood were measured after HVT ventilation (#p<0.05, ##p<0.01, ###p<0.001 vs NV, *p<0.05, **p<0.01; NV n=8, LVT n=13, LVT+AM n=12, HVT n=10, HVT+AM n=10).
AM prevented VILI-associated elevation of blood lactate levels
HVT significantly increased blood lactate levels after 2 h of ventilation, suggesting tissue hypoperfusion and impairment of microcirculation. Blood lactate levels were normalised by AM infusion (figure 5A).

Adrenomedullin (AM) prevented ventilator-induced lung injury (VILI)-associated disturbance of the systemic microcirculation. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT); intervention groups were treated with AM. HVT ventilation induced an increase of lactate plasma levels, indicating disturbance of the systemic microcirculation absent in LVT-ventilated mice. AM attenuated the increase of lactate levels during HVT ventilation (**p<0.01, ***p<0.001; LVT n=14, LVT+AM n=12, HVT n=11, HVT+AM n=9).
Destabilisation of the macrocirculation did not contribute to the raise of blood lactate levels under HVT. Mean arterial blood pressure (MAP) was >70 mm Hg at any time in all experimental groups and tended to be lower in the LVT groups as compared with HVT groups. Further, AM is known to be a potent vasodilator, and AM treatment caused an initial depression of MAP in the treated as compared with the untreated HVT group. Within 1 h of AM treatment, the difference in MAP between the groups subsided. (online data supplement, figure 1).
AM treatment attenuated VILI-associated renal dysfunction
Injurious ventilation is known to affect distal organ function and has been linked to renal failure.16 20 21 In our model, HVT ventilation evoked an increase of creatinine levels as compared with LVT-ventilated or non-ventilated mice, suggesting renal dysfunction (figure 6). Treatment with AM prevented the rise of creatinine levels during HVT ventilation (figure 6). AST and ALT levels were not altered by MV. However, ALT levels were elevated in AM-treated mice of the HVT group as compared with either untreated HVT-ventilated mice or AM-treated LVT-ventilated mice (online data supplement, figure 2).

Adrenomedullin (AM) treatment attenuated ventilator-induced lung injury (VILI)-associated renal dysfunction. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT); intervention groups were treated with AM (NV, non-ventilated mice). HVT ventilation led to an increase of creatinine levels compared with LVT-ventilated and non-ventilated mice, indicating renal failure. Treatment with AM significantly prevented the rise of creatinine levels during HVT ventilation (***p<0.001; n=6).
AM treatment did not significantly alter pulmonary and systemic cytokine production
LVT and HVT increased the production of IL-6, KC and MIP-2 in the lung as compared with non-ventilated controls (figure 7A). The levels of IL-6, KC and MIP-2 were not significantly reduced in the HVT group treated with AM as compared with the HVT control group. Both ventilatory modes evoked a systemic inflammatory response, with increased IL-6 and MIP-1α in plasma (figure 7B). Levels of investigated cytokines in the systemic circulation were not affected by treatment with AM in any of the ventilated groups.

Pulmonary and systemic cytokine production. Mice were ventilated with tidal volumes of 7 ml/kg (LVT) or 17 ml/kg (HVT); intervention groups were treated with adrenomedullin (AM) (NV, non-ventilated mice). (A) LVT and HVT elicited the production of interleukin 6 (IL-6), keratinocyte-derived chemokine (KC) and macrophage-inflammatory protein-2 (MIP-2) in the lung compared with non-ventilated controls. AM had no impact on pulmonary cytokine levels measured in bronchoalveolar lavage fluid (BALF) (*p<0.05, **p<0.01, ***p<0.001 vs NV; n=6). (B) Mechanical ventilation resulted in a rise of IL-6, MIP-1α and MIP-2 levels unaffected by AM treatment (**p<0.01, ***p<0.001 vs NV; #p<0.05, n=6).
Infusion of AM attenuated VILI after 6 h of MV when started after 2 h of MV
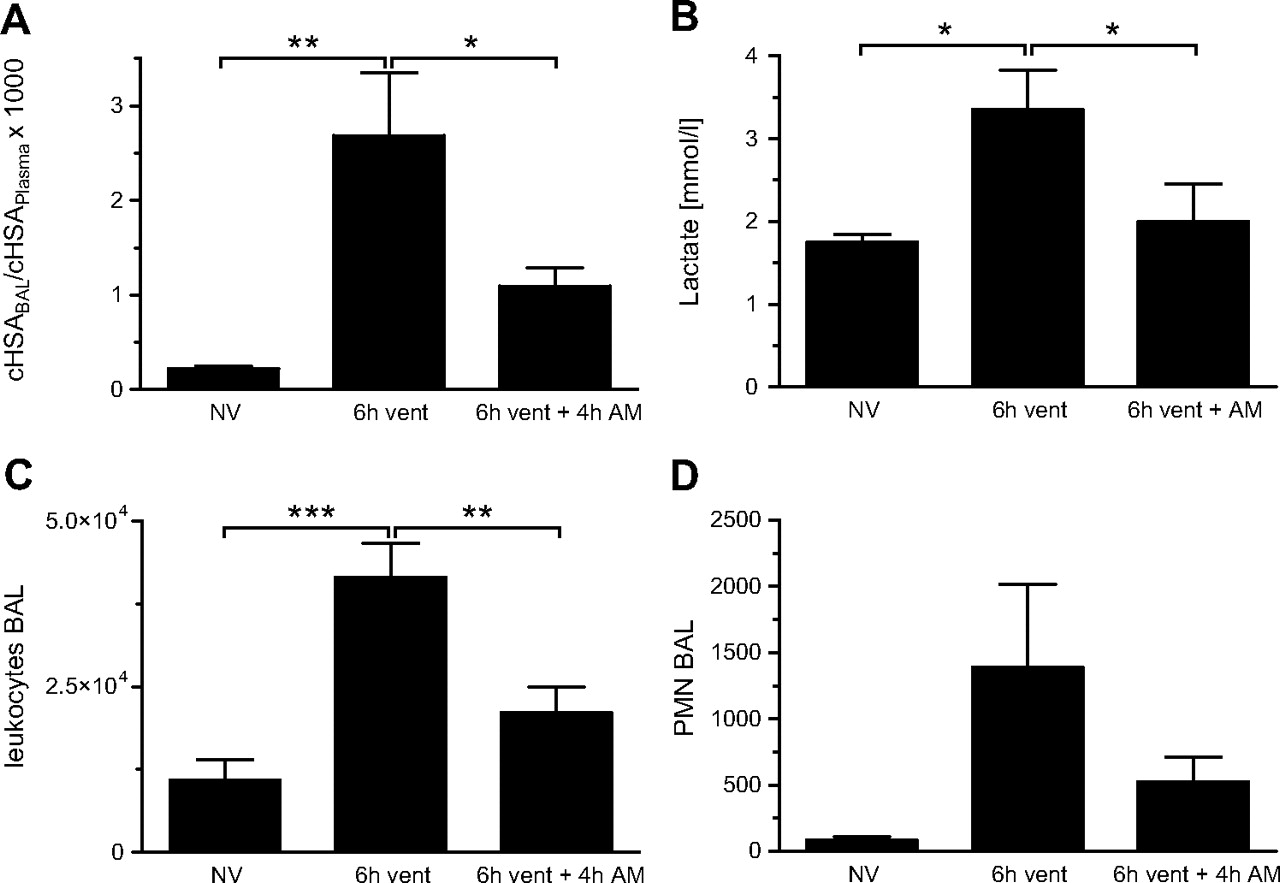
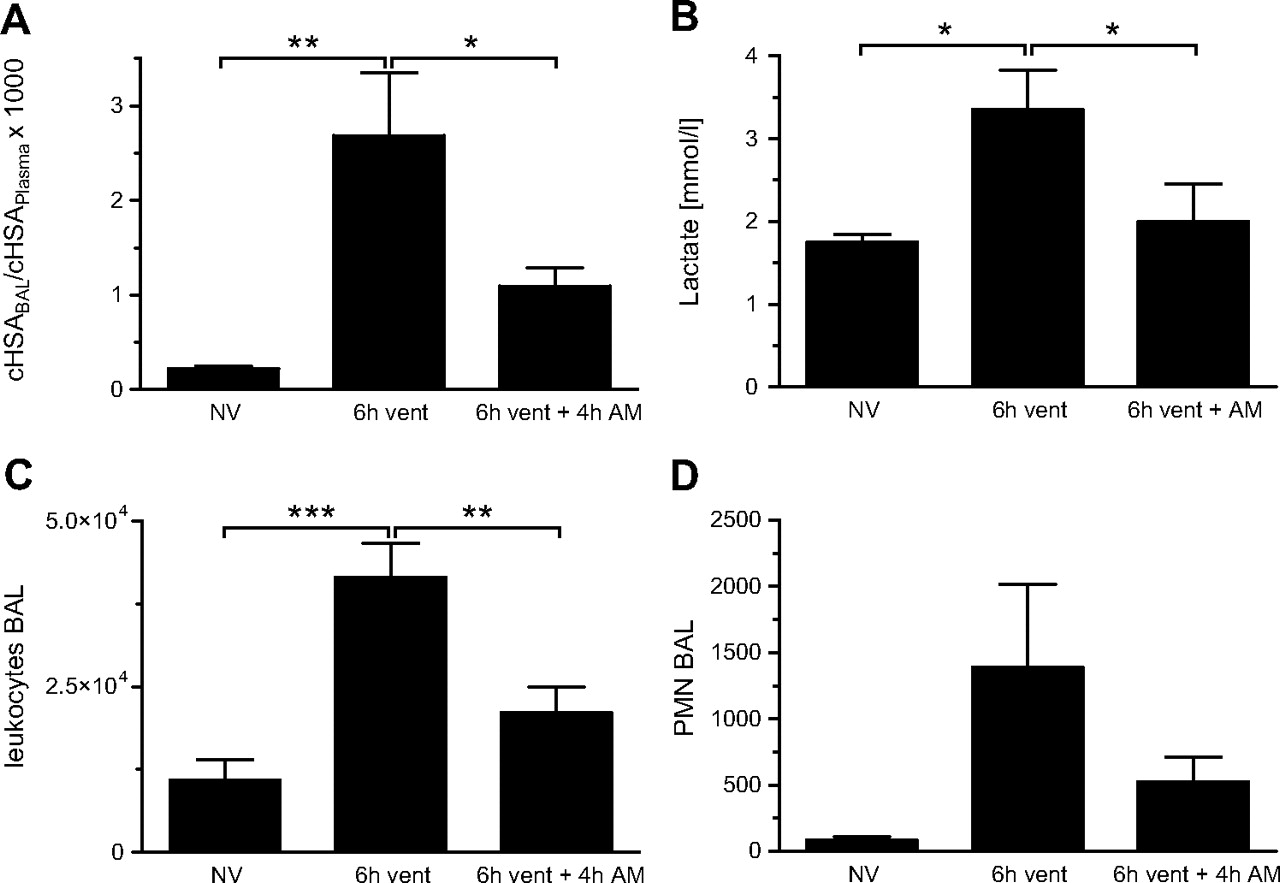
To evaluate whether AM exerts beneficial effects on VILI in a therapeutic setting, mice were ventilated for 6 h, and AM treatment was started 2 h after the induction of MV. MV provoked a marked increase of lung vascular permeability and increased blood lactate levels after 6 h of MV, suggesting tissue hypoperfusion and impairment of microcirculation. The numbers of leucocytes and particularly PMNs in BALF were increased by MV. Systemic application of AM attenuated pulmonary hyperpermeability (figure 8A), normalised blood lactate levels (figure 8B) and limited invasion of the lung by leucocytes (figure 8C) and neutrophils (figure 8D) even when AM infusion was started 2 h after the initiation of MV.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adrenomedullin (AM) attenuated ventilator-induced lung injury (VILI) after 6 h of mechanical ventilation (MV) when being started after 2 h of MV. Mice were ventilated with a tidal volume (VT) of 12 ml/kg for 6 h (6 h vent). Adrenomedullin was infused for 4 h starting 2 h after initiation of MV (6 h vent+4 h AM) (NV, non-ventilated mice). (A) Human serum albumin (HSA) was infused 90 min prior to termination of the experiment. An increased HSA bronchoalveolar lavage (BAL)/plasma ratio indicated microvascular leakage. AM treatment reduced pulmonary vascular leakage evoked by MV (*p<0.05, **p<0.01; n=5). (B) 6 h of MV induced an increase of plasma lactate levels, indicating disturbance of the systemic microcirculation, which was attenuated by therapeutic AM infusion (*p<0.05, n=5). Further, pulmonary leucocyte (C) and polymorphonuclear leucocyte (PMN) (D) influx was reduced by AM in this model (**p<0.01, ***p<0.001; n=5).
Discussion
AM attenuated lung hyperpermeability, MLC phosphorylation, leucocyte recruitment to the alveolar space, deterioration of the systemic microcirculation and renal dysfunction in ventilated mice. Therapeutic effects were more pronounced in the HVT group than in the LVT group and were evident even when AM treatment was initiated 2 h after MV in a model of 6 h ventilation. By employing different mouse models of MV, the current study provides evidence that continuously infused AM might limit VILI.
Mouse models have been successfully used to investigate the pathobiology of VILI and provided knowledge on central pathomechanisms in MV.22 However, some challenges of VILI mouse models need to be considered. LVT ventilation requires high respiratory rates to eliminate CO2 sufficiently in mice, possibly resulting in endogenous PEEP, which may contribute to lung injury.23 Further, the observation period is usually limited to a few hours as compared with ventilation of humans for days or weeks. In addition, most experimental studies on VILI employed lungs of healthy mice instead of preinjured lungs. In the current study, an LVT protocol of 7 ml/kg was used to reproduce the low tidal ventilation strategy recommended for human patients as accurately as possible. Of note, the respiration rate of 240/min which was required to avoid alveolar hypoventilation is within the reported physiological range for C57Bl/6 mice,24 and monitoring of ventilation slope and airway pressure did not suggest relevant endogenous PEEP. Further, whereas the current study did not employ preinjured lungs, a ‘second hit’ was implemented by ventilating with pure oxygen, which is known to be noxious but frequently required in patients with ARDS. In addition to LVT, HVT (17 ml/kg) was employed, which induced moderate lung inflammation and permeability, thus displaying improved suitability for analysing protective AM-evoked effects within the observation period of 2 h. Besides overinflation, atelectrauma, which was reduced by PEEP in the LVT groups, presumably contributed to the development of VILI in the HVT groups. Further, a 6 h model of VILI employing a tidal volume of 12 ml/kg was established, which allowed observation for a longer period of time. Respiratory rates could be reduced to 120/min due to a tidal volume of 12 ml/kg in this model. According to the baby lung concept of inhomogeneously injured ARDS lungs, lung stress and lung strain generated by a tidal volume of 12 ml/kg in healthy lungs may apply in ventilated areas of inhomogeneously injured lungs even under lung-protective ventilation, which underscores the clinical relevance of the current experimental approach.25 26 To enhance the clinical relevance of the model further, haemodynamic instability was prevented by fluid support.
Impairment of pulmonary endothelial barrier function is a hallmark of VILI, leading to the development of lung oedema, surfactant dysfunction and deterioration of pulmonary gas exchange.3 Disruption of the pulmonary endothelial barrier may result from endothelial cell contraction and interendothelial gap formation evoked by myosin–actin interaction following MLC phosphorylation.19 Notably, MLC phosphorylation contributed to the development of lung hyperpermeability in ALI and VILI.27 28 AM protected against vascular hyperpermeability in lung, kidney, ileum and liver in experimental models of sepsis or ALI,9 11–13 and inhibition of MLC phosphorylation was recognised as a major underlying mechanism in the current study. AM reduced VILI-associated pulmonary hyperpermeability, which was accompanied by diminished MLC phosphorylation. It is tempting to speculate that AM-evoked reduction of MLC phosphorylation may have been contributing to improved lung endothelial barrier function. Moreover, we observed improved oxygenation in AM-treated mice subjected to VILI. Although the P/F ratio was only moderately changed, the significant oxygenation improvement in the AM-treated group after only 2 h of MV underscores the functional relevance of the current observations.
Leucocytes and particularly PMNs contribute to microvascular permeability and tissue damage in VILI.29 Leucocyte transmigration from blood into lung tissue depends on chemotactic gradients, as well as mechanisms requiring tightly regulated leucocyte–endothelial interplay that include expression of adhesion molecules on both cell types.30 Of note, AM reduced CD11b expression on activated human PMNs in vitro, which is crucial for their attachment to endothelial cells and tissue transmigration in VILI.30 31 In isolated rat ileum, AM prevented leucocyte attachment to capillaries following stimulation with Staphylococcus aureus α-toxin.14 In line with these results, AM reduced the VILI-associated rise in alveolar PMNs in the present study. In parallel, the increase of circulating blood PMNs following HVT ventilation in AM-treated mice was higher as compared with HVT-ventilated and untreated mice. Although not directly evidenced by the current study, the previous and current data together suggest that AM-induced impairment of leucocyte–endothelial interaction may have been contributing to diminished PMN recruitment into the alveolar compartment. In addition, inflammatory cytokines were increased in BALF and plasma of mice ventilated with HVT or LVT for 2 h in the current investigation. Even a protective ventilation strategy in the absence of hyperoxia has been reported to cause significant liberation of inflammatory cytokines in mice as well as in humans2 32; thus increased liberation of proinflammatory cytokines even in the LVT group could be expected. Of note, the currently analysed cytokine levels were not reduced by AM treatment, suggesting that improvement of barrier function, reduction of leucocyte infiltration and attenuation of systemic effects including increases in lactate concentrations were not secondary to reduced levels of these cytokines.
Systemic hyperinflammation drives deterioration of the microcirculation and may cause renal failure.33 34 Kidneys are usually the first organ system to be involved in VILI-associated multiorgan failure in humans, and AM reportedly improved renal performance in septic shock in rats and in human congestive heart failure.13 16 35 In the current investigation, HVT ventilation evoked elevated plasma creatinine levels, suggesting renal dysfunction, and AM treatment prevented this increase of creatinine. AST and ALT as surrogate markers of hepatic injury were not altered by any MV protocol. However, exclusively under AM treatment ALT levels were elevated in HVT-ventilated animals. Although no direct association between AM treatment and hepatic injury has been reported yet, this finding warrants critical evaluation in further studies. Of note, a considerable increase in blood lactate was observed in the present study in HVT-ventilated, untreated mice, which may suggest impaired microcirculation. AM prevented blood lactate increase under MV-induced systemic inflammatory conditions. Reportedly, AM prevented the development of microcirculatory damage in isolated rat ileum, and protection against endothelial leucocyte adhesion contributed to this beneficial effect.11
The current data suggest that AM may limit VILI in the presence of hyperoxia. Since mechanical ventilation is indispensable in patients with ALI and ARDS, VILI may contribute to poor clinical outcome even when lung-protective ventilation strategies are applied. Of note, clinical application of AM has been reported to be beneficial and safe in congestive heart failure.35 To this regard, the potential translation of AM into clinical trials is considerably stimulated by a recent statement of the EMA which recommends the development of AM as orphan drug for patients with ALI (EMA/COMP/104704/2010).
In conclusion, AM reduced pulmonary vascular permeability and improved oxygenation in different mouse models of hyperoxic MV, even when being infused after the induction of lung injury by MV. Further, leucocyte recruitment to the alveolar space was diminished by AM treatment. AM also ameliorated the ventilation-evoked increase of lactate and creatinine levels, probably indicating improvement of the systemic microcirculation and renal function, respectively. The current data suggest that AM may provide a promising perspective as adjuvant treatment to limit VILI in addition to lung-protective ventilation. The recent positive recommendation of the EMA regarding the clinical development of AM for patients with ALI further underscores the clinical relevance of the current experimental study.
Acknowledgments
The excellent technical assistance of J. Hellwig and K. Hellwig is greatly appreciated. This work contributes to the MD thesis of HCM.
References
Supplementary materials
Web Only Data thx.2010.135996
Files in this Data Supplement:
Footnotes
Funding Supported in part by grants from the German Research Foundation to MW (OP 86/7-1 and SFB/TR 84, C3 and C6) and SH (HI-789/6-1), and the German Federal Ministry of Education and Research to HCM, SH, NS and SR (Pneumonia Research Network on Genetic Resistance and Susceptibility for the Evolution of Severe Sepsis, PROGRESS).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.