Article Text

Abstract
Background Infections with some bacteria, including Streptococcus pneumoniae, have been associated with a reduced incidence of asthma. Components of S pneumoniae may have the potential to modulate allergic inflammatory responses and suppress the development of asthma.
Objectives To determine if human S pneumoniae vaccines have the potential to suppress asthma by elucidating their effect on allergic airways disease (AAD) in mouse models.
Methods AAD was induced in BALB/c mice by intraperitoneal sensitisation and intranasal challenge with ovalbumin. Pneumococcal conjugate or polysaccharide vaccines were administered at the time of sensitisation or during established AAD. Hallmark features of AAD were assessed. Levels of regulatory T cells (Tregs) were quantified by fluorescence-activated cell sorting, and their immunoregulatory capacity was assessed using proliferation assays and anti-CD25 antibody treatment.
Results Intranasal administration of the conjugate vaccine, but not the polysaccharide vaccine, suppressed the hallmark features of AAD, including: eosinophilic and T helper 2-mediated inflammation; airway hyper-responsiveness; circulating immunoglobulin E (IgE) levels; and mucus hypersecretion. Intramuscular administration of the conjugate vaccine had limited protective effects. The conjugate vaccine increased Tregs in the lung-draining lymph nodes, lung and spleen. Furthermore, conjugate vaccine-induced Tregs had an enhanced capacity to suppress T effector responses. Anti-CD25 administration reversed the suppressive effects of the conjugate vaccine.
Conclusions A currently available human conjugate vaccine suppresses the hallmark features of AAD through the induction of Tregs. Thus targeted administration may provide a novel immunoregulatory treatment for asthma.
- Asthma
- Streptococcus pneumoniae
- regulatory T cells
- immunoregulatory therapy
- allergic lung disease
- asthma mechanisms
Statistics from Altmetric.com
- Asthma
- Streptococcus pneumoniae
- regulatory T cells
- immunoregulatory therapy
- allergic lung disease
- asthma mechanisms
Introduction
The prevalence of asthma varies inversely with the incidence of certain bacterial infections such as tuberculosis and typhoid.1 Furthermore, exposure to bacterial DNA motifs, such as CpG-oligodeoxynucleotides (ODNs), suppresses allergic inflammation by inducing a T helper 1 (Th1) response in mouse models of asthma.2 However, utilisation of Th1-inducing agents to suppress allergic airways disease (AAD) has not translated well into clinical practice.
The development and progression of allergy and asthma may result from a lack of infection-induced regulatory T cell (Treg) expansion that leads to maladaptive immune responses and the development of disease. Indeed, several studies have linked impaired or altered Tregs with asthma.3–7 Many infectious agents induce Tregs and protect against AAD in murine models.8 The success of Treg-driven suppression of AAD is due to their capacity to inhibit multiple effector arms of the immune responses involved in AAD. This includes attenuation of the function of Th2 cells, macrophages, dendritic cells (DCs), natural killer T cells and B cells. Hence, suppression of allergic inflammation by the induction of Treg responses may provide a mechanism for the suppression of disease.
Utilisation of immunoregulatory treatments that are based on bacterial components, which modulate allergic responses through Treg induction, may have benefits as therapeutic strategies for the suppression of asthma.8 Tregs are crucial in protection against Streptococcus pneumoniae infection and are induced to substantially greater levels compared with other Gram-positive bacteria.9 Streptococcus pneumoniae vaccination reduced the incidence of asthma and associated hospitalisations in both children and the elderly.10 11 Furthermore, S pneumoniae infection was associated with reduced eosinophilia and AAD in animal models.12 13 In addition, we have shown that killed S pneumoniae suppresses features of AAD in mouse models.14 These studies suggest that there is an opportunity for the development of S pneumoniae-based immunoregulatory treatments for asthma.15
A major hurdle in the development of successful immunoregulatory treaments for asthma, based on infectious agents, is the identification of effective suppressive factors. In this study we used S pneumoniae-based vaccines, which are currently used in humans, to determine their efficacy in suppressing AAD in mouse models. Treatment with the conjugate vaccine attenuated the expression of hallmark features of AAD through the expansion of the Treg pool.
Methods
See the online supplement for additional details.
Mice
Female 6- to 8-week-old BALB/c mice were obtained from the Animal Facility. All procedures were approved by the Animal Ethics Committee, University of Newcastle.
AAD models
To induce AAD, mice were sensitised to ovalbumin (OVA; intraperitoneal day 0; 50 μg; Sigma-Aldrich, St Louis, Missouri, USA) with Rehydrogel (1 mg; Reheis, Berkeley Heights, New Jersey, USA) in saline (200 μl) and challenged (OVA, intranasal days 12–15; 10 μg/50 μl saline, figure 1A).14 To recapitulate established disease, mice received OVA intraperitoneally (day 0) followed by two challenge periods (days 11–13 and 32–34; figure 2A). AAD was assessed 24 h after the final challenge.

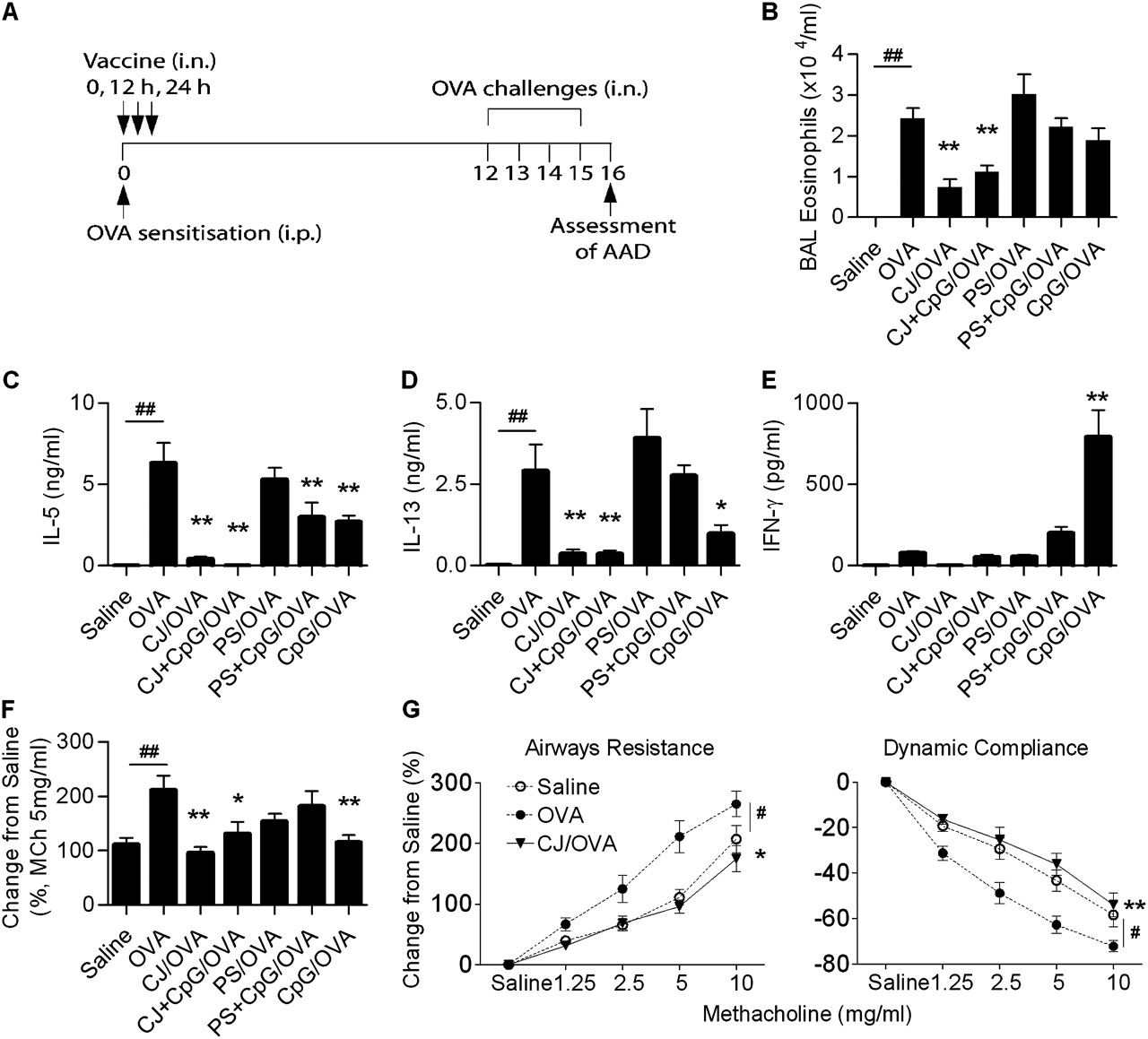
(A) Experimental protocol to assess the effect of vaccines on the induction of allergic airways disease (AAD). AAD was induced by intraperitoneal (i.p.) ovalbumin (OVA) sensitisation and intranasal (i.n.) OVA challenge. Controls received saline sensitisation and OVA challenge. Vaccine treatment was administered at the time of OVA sensitisation, in three doses every 12 h. Features of AAD were assessed 24 h after completion of the OVA challenge. The effect of conjugate (CJ) and polysaccharide (PS) vaccines, with and without CpG-oligodeoxynucleotides (ODNs), on (B) eosinophils in bronchoalveolar lavage (BAL), OVA-induced (C) interleukin 5 (IL-5), (D) IL-13 and (E) interferon γ (IFNγ) release from mediastinal lymph node (MLN) T cells and (F) airways resistance at the representative dose of 5 mg/ml methacholine (MCh) reflective of full dose–response curves. The effect of CJ on airway hyper-responsiveness is also shown as (G) full dose–response curves for airways resistance and dynamic compliance. Data represent the mean±SEM from 6–8 mice. Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as #p<0.05, ##p<0.01. Significant differences between OVA-sensitised and vaccine-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01. Significant differences for resistance and compliance are for the entire dose–response curves.
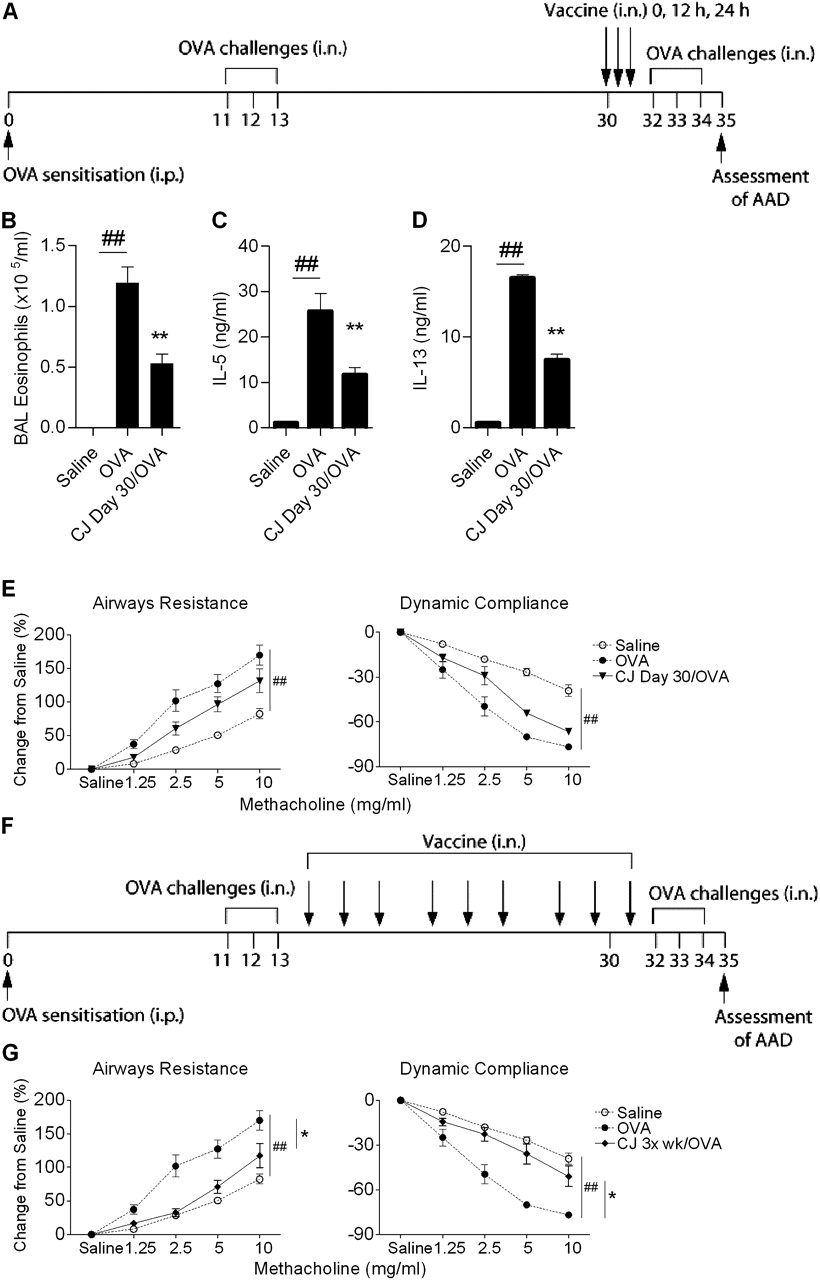
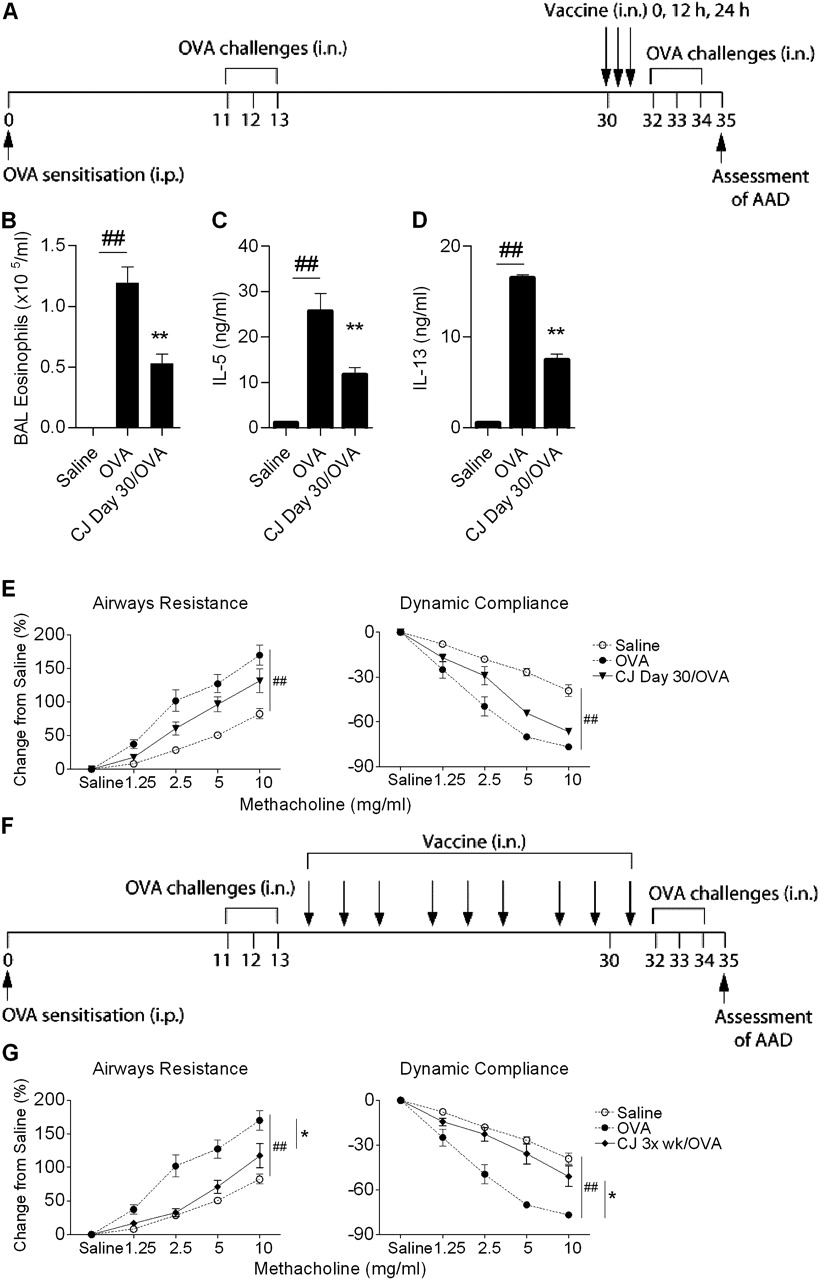
(A) Experimental protocol to assess the effect of conjugate vaccine (CJ) treatment on established allergic airways disease (AAD). AAD was established by intraperitoneal (i.p.) ovalbumin (OVA) sensitisation followed by an initial intranasal (i.n.) OVA challenge and then a second challenge to recapitulate AAD in established disease. CJ was administered on day 30, in three doses every 12 h. The effect of CJ treatment on (B) eosinophils in bronchoalveolar lavage (BAL), OVA-induced (C) interleukin 5 (IL-5) and (D) IL-13 release from mediastinal lymph node (MLN) T cells and (E) airway hyper-responsiveness (AHR). (F) The effect of CJ treatment delivered three times per week commencing on day 14 (totalling nine doses) on (G) AHR. Data represent the mean±SEM from 6–8 mice. Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as #p<0.05, ##p<0.01. Significant differences between OVA-sensitised and CJ-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01. Significant differences for resistance and compliance are for the entire dose–response curves.
Vaccine treatment
Mice were administered (33 μl) 7-valent polysaccharide conjugate (Wyeth, Madison, New Jersey, USA) or 23-valent polysaccharide vaccine (Merck, Whitehouse Station, New Jersey, USA), ±CpG-ODN (10 μg, TCCATGACGTTCCTACGTT; Geneworks, Thebarton, SA, Australia), intranasally every 12 h for three doses. To assess the effects of the conjugate vaccine in established AAD, mice were treated one or three times per week (figure 2). To assess administration by a different route, conjugate vaccine was delivered intramuscularly. Where indicated, mice received anti-CD25 intraperitoneally (day–3, 100 μg/200 μl saline, PC61; eBioscience, San Diego, California, USA).
Assessment of cellular inflammation
Differential cell counts of May Grunwald–Giemsa-stained bronchoalveolar lavage (BAL) were enumerated.16
T cell cytokines
Cytokine release by T cells restimulated with OVA was determined in supernatants by ELISA.14
Airways hyper-responsiveness (AHR)
AHR was assessed by restrained invasive plethysmography as previously described.16
Serum antibodies
Total immunoglobulin E (IgE) and OVA-specific IgG1 (using OVA 2 mg/well as the capture antigen) concentrations were determined by ELISA.13
Lung histology
Lung sections were stained with periodic acid–Schiff or chrome salt fixation, and airway mucus-secreting cells and tissue eosinophils were determined.16
Flow cytometry
Single cell suspensions were stained for surface-associated CD4, CD25, CD103, cytotoxic T lymphocyte antigen 4 (CTLA-4; BD Pharmingen, San Diego, California, USA) and transforming growth factor β1 (TGF β1; R&D Systems, Minneapolis, USA) then fixed, permeabilised and stained for intracellular FoxP3 (eBioscience) and analysed by flow cytometry (FACS Canto, BD Biosciences, Mississauga, Canada).13
Cell isolation
CD4+CD25+ cells were isolated (>90% pure) using CD11c depletion followed by a CD4+CD25+ Treg isolation (AutoMACs Pro, Miltenyi Biotec, Auburn, California, USA). CD4+CD25– cells were further purified by CD4+ selection (>94% pure).13
Cell suppression assay
CD4+CD25– cells (5×104) and varying numbers of CD4+CD25+ cells were cultured in RPMI (200 μl, 10% fetal calf serum; 72 h, 37°C) with anti-CD28 (1 μg/ml; BD Pharmingen) and OVA (1 μg/ml) in 96-well plates. Cells were pulsed for the final 18 h of culture with [3H]thymidine (Amersham International, Amersham, UK) and enumerated using a microbeta counter (Wallac TriLux, PerkinElmer, Waltham, Massachusetts, USA).13
Data analysis
Data were analysed using GraphPad Prism (GraphPad Software, California, USA) and are represented as the mean±SEM. One-way analysis of variance (ANOVA) with the Dunnett post-test was used to determine significance between data with multiple comparisons. One-way repeated measures ANOVA and the Bonferroni post-test were used to determine significance for AHR data. Unpaired Student t test was used to determine differences between the two groups. A p value <0.05 was considered statistically significant. Where p is between 0.01 and 0.05 the numerical value is provided.
Results
Treatment with the conjugate but not polysaccharide vaccine suppresses the development of hallmark features of AAD
We first investigated the effect of pneumococcal vaccines on the development of AAD. Mice were treated with either the conjugate or polysaccharide vaccine, with or without CpG-ODN (as a positive control and potential adjuvant), at the time of OVA sensitisation, and the effect of vaccine treatment on hallmark features of AAD was assessed (figure 1A).
Airway challenge of OVA-sensitised mice resulted in the induction of AAD, which was characterised by increased numbers of eosinophils in BAL fluid, OVA-induced interleukin 5 (IL-5) and IL-13 release from mediastinal lymph node (MLN) T cells, and AHR (figure 1B–G). AAD was not induced in saline-sensitised OVA-challenged mice (figure 1B–G).
Treatment of OVA-sensitised mice with the conjugate vaccine suppressed the development of hallmark features of AAD, including eosinophil numbers in BAL fluid, OVA-induced IL-5 and IL-13 release from MLN T cells, and AHR (figure 1A–G). The administration of CpG-ODN had no additional effect. In contrast, treatment with polysaccharide vaccine had no effect on AAD (figure 1A–F; see Supplementary figure 1 for dose–response curves). Administration of the polysaccharide vaccine with CpG-ODN attenuated OVA-induced IL-5 and IL-13 release to the same level as CpG-ODN alone, suggesting that the preventive effects were due to CpG-ODN. CpG-ODN treatment also suppressed the development of AHR, IL-5 and IL-13 (p=0.0361), but did not alter BAL eosinophil numbers. Unlike treatment with the conjugate vaccine, attenuation of AAD by CpG-ODN was associated with a substantial increase in OVA-induced interferon γ (IFNγ) release from MLN T cells (figure 1E).
Conjugate vaccine treatment before or after sensitisation suppresses the development of AAD
In addition to treatment at the time of sensitisation, the conjugate vaccine suppressed the development of AAD when administered before or after OVA sensitisation (Supplementary figure 2). These data suggest that the effects of treatment persist and last for at least 26 days. The backbone components (AlPO4+CRM197) of the conjugate vaccine did not suppress the development of AAD (Supplementary figure 3).
Conjugate vaccine treatment suppresses established AAD
We then investigated whether administration of the conjugate vaccine would be effective in established AAD. A model of AAD was developed that involved sensitisation to OVA, followed by initial challenges (days 11–13) to establish AAD and second challenges (days 32–34) to recapitulate disease before the assessment of AAD (figure 2A). The conjugate vaccine was administered during established AAD (day 30). The conjugate vaccine suppressed eosinophils in BAL fluid and OVA-induced IL-5 and IL-13 release from MLN T cells (figure 2B–D). However, this regime did not suppress AHR in a statistically significant manner (figure 2E). Therefore, the number of doses was increased, and treatment with the conjugate vaccine three times per week (commencing on day 14) suppressed AHR (figure 2G).
Intramuscular administration of the conjugate vaccine had limited effects on the development of AAD
The conjugate vaccine is currently administered to humans to prevent S pneumoniae infections via the intramuscular route. Therefore, the effect of intramuscular administration of the conjugate vaccine on the development of AAD was investigated (Supplementary figure 4A). Intramuscular administration suppressed eosinophil numbers in BAL fluid and OVA-induced IL-5 and IL-13 (p=0.0312) release from splenic T cells; however, neither IL-5 or IL-13 release from MLN T cells nor AHR were suppressed (Supplementary figure 4B–G).
Conjugate vaccine treatment suppresses the development of systemic Th2 responses
We then investigated the effects of intranasal administration of the conjugate vaccine on systemic Th2 responses, antibody responses and airway inflammation. The conjugate vaccine suppressed OVA-induced IL-5 and IL-13 cytokine release from splenic T cells, circulating levels of IgE (p=0.0227) and OVA-specific IgG1 in serum, reduced the numbers of mucus-secreting cells and eosinophils in airway tissue (within 100 μm of the surface) and suppressed the number of circulating eosinophils (figure 3A, B)

The effect of conjugate vaccine (CJ) treatment on ovalbumin (OVA)-induced (A) interleukin 5 (IL-5) and (B) IL-13 release from splenocyte T cells, serum levels of (C) total immunoglobulin E (IgE) and (D) OVA-specific IgG1, numbers of (E) mucus-secreting cells (MSCs), numbers of (F) eosinophils in the airway tissue, and (G) eosinophils in blood. Arrows indicate MSCs or eosinophils in airway tissue. Data represent the mean±SEM from 6–8 mice. Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as ##p<0.01. Significant differences between OVA-sensitised mice and CJ-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01.
Conjugate vaccine treatment induces Tregs
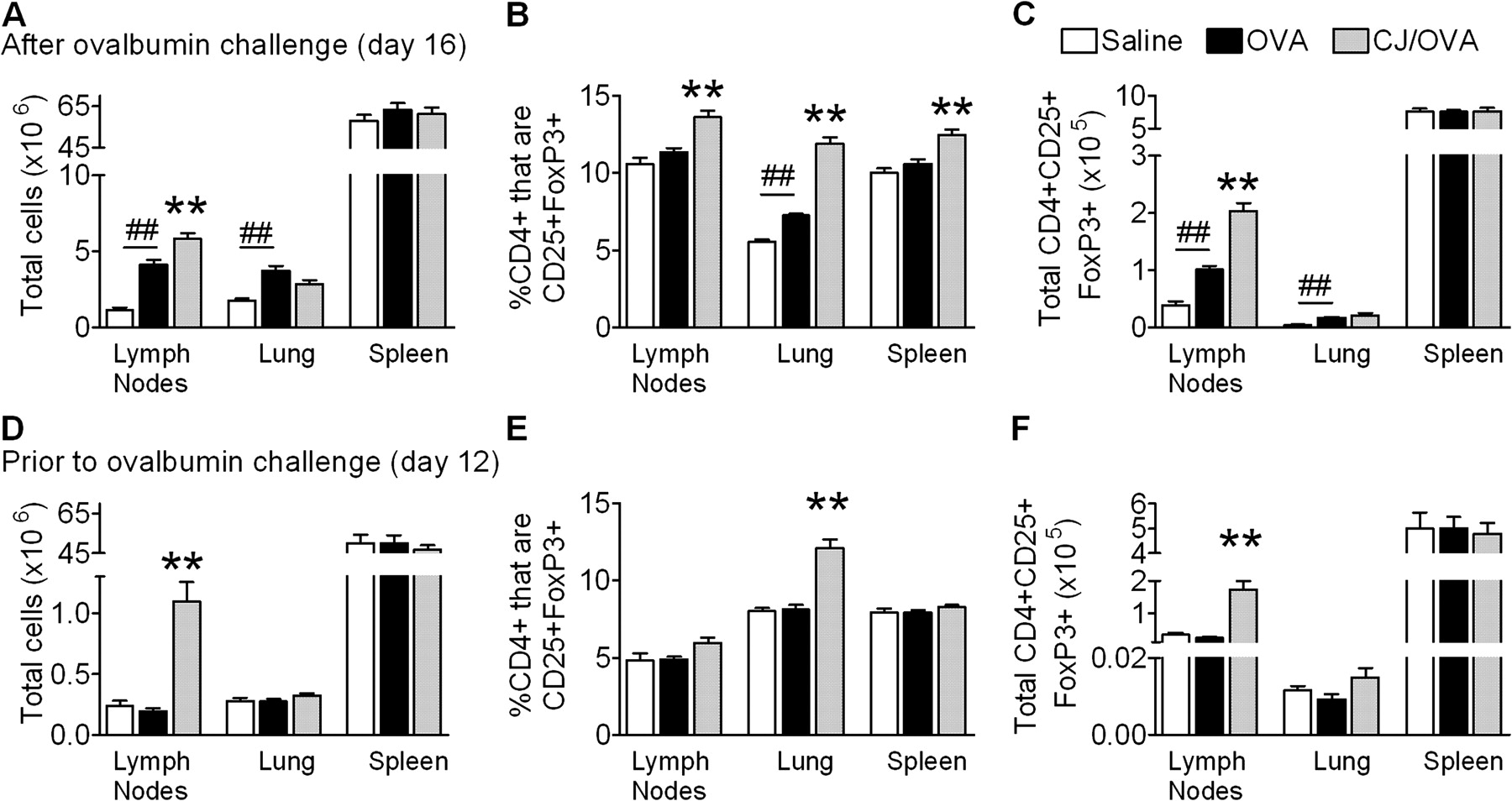
The suppression of AAD by the conjugate vaccine was not associated with an increased release of T cell-derived IFNγ in the MLNs. Therefore, we examined the role of Tregs in conjugate vaccine-mediated suppression of AAD. The number of CD4+CD25+FoxP3+ Tregs in the MLNs, lungs and spleens was assessed after the induction of AAD (day 16). Conjugate vaccine-treated OVA-sensitised mice had significant increases in the total number of cells in the MLNs compared with untreated OVA-sensitised mice (figure 4A). The percentage of CD4+ cells that were CD25+FoxP3+ in the MLNs, lung and spleen and the total number of CD4+CD25+FoxP3 cells in the MLNs were also increased (figure 4B, C).
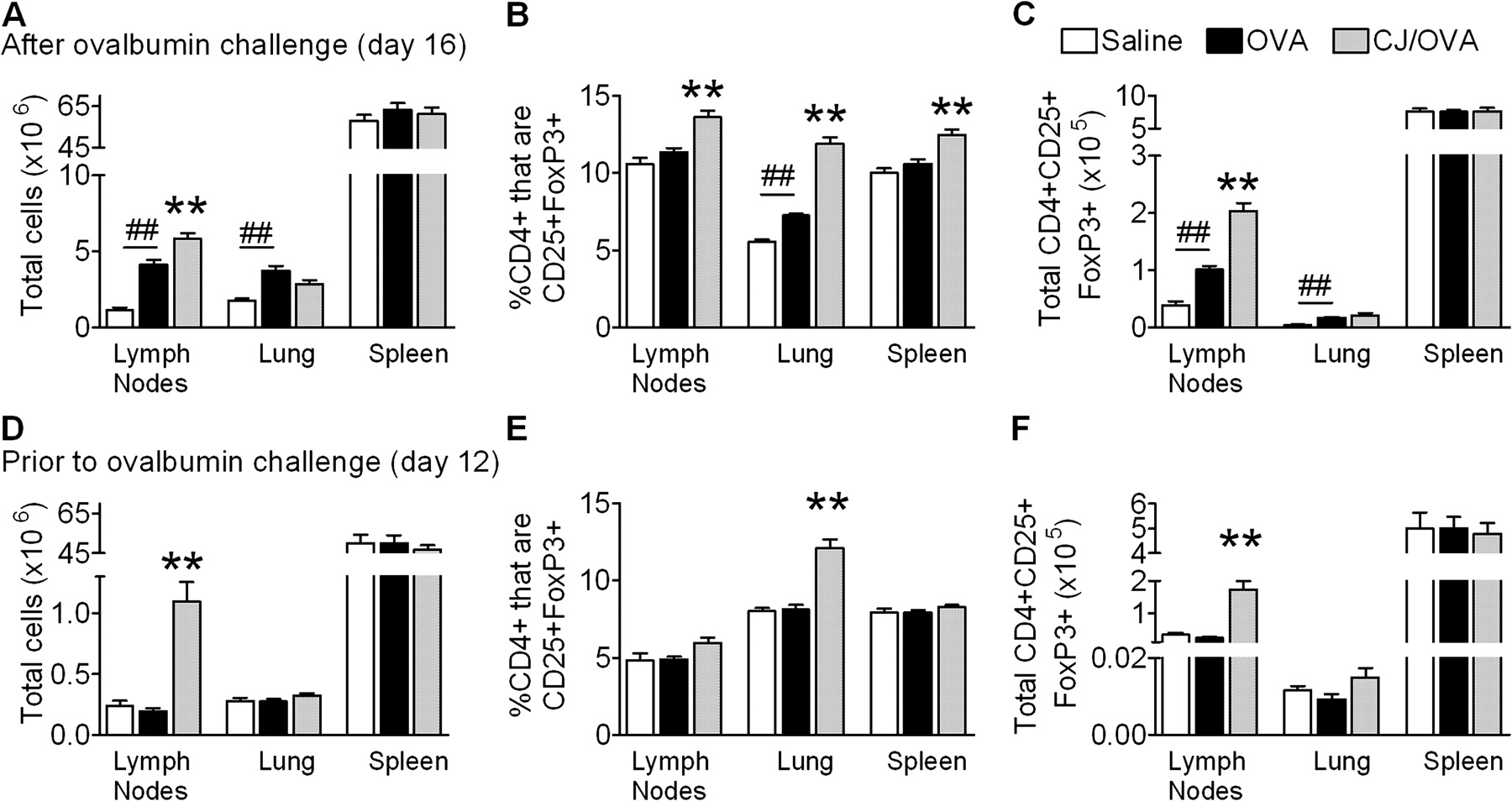
The effect of conjugate vaccine (CJ) treatment on (A) total number of cells, (B) percentage of CD4+ cells that are CD25+FoxP3+ and (C) total CD4+CD25+FoxP3+ cells in the mediastinal lymph nodes (MLNs), lung and spleen in allergic airways disease. The effect of CJ treatment, assessed prior to ovalbumin (OVA) challenge on (D) total number of cells, (E) percentage of CD4+ cells that are CD25+FoxP3+ and (F) total CD4+CD25+FoxP3+ cells in the MLNs, lung and spleen. Data represent the mean±SEM from 6–8 mice. Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as ##p<0.01. Significant differences between OVA-sensitised mice and CJ-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01.
We then investigated whether conjugate vaccine treatment induced CD4+CD25+FoxP3+ cells prior to OVA challenge. Again, treatment increased the total number of cells in MLNs (figure 4D). The percentage of CD4+CD25+ cells that were FoxP3+ and the total number of CD4+CD25+FoxP3+ cells in the MLNs and lung were also increased (figure 4E, F).
Conjugate vaccine treatment induces Tregs that have a greater suppressive capacity
IL-10 and TGFβ1 are immunosuppressive cytokines that may mediate the suppressive effects of Tregs. Therefore, the effect of conjugate vaccine treatment on the levels of expression of OVA-induced IL-10 and TGFβ1 release from MLN T cells was determined. Treatment suppressed OVA-induced IL-10 and TGFβ1 (p=0.0224) release from MLN T cells compared with untreated OVA-sensitised mice (figure 5A, B). The percentage of CD4+CD25+ cells expressing membrane-bound TGFβ1 remained unchanged (figure 5C). Representative flow cytometric data are provided (Supplementary figure 5).

The effect of conjugate vaccine (CJ) treatment on ovalbumin (OVA)-induced (A) interleukin 10 (IL-10) and (B) transforming growth factor β (TGFβ) release from mediastinal lymph node (MLN) T cells, the percentage of CD4+CD25+FoxP3+ cells expressing (C) membrane-bound TGFβ (mTGF-β), (D) CD103 and (E) cytotoxic T lymphocyte antigen 4 (CTLA-4), and (F) the percentage of CD4+CD25+ that are FoxP3+. (G) Proliferation of CD4+CD25– cells cultured with varying numbers of CD4+CD25+ Tregs from OVA-sensitised or CJ-treated OVA-sensitised mice and stimulated with OVA and anti-CD28. Data represent the mean±SEM from 6–8 mice (A–F) or triplicate wells (G). Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as #p<0.05, ##p<0.01. Significant differences between OVA-sensitised mice and CJ-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01.
The levels of other markers associated with the suppressive activity of Tregs were also assessed. Conjugate vaccine treatment increased the expression of CD103 and CTLA-4 (p=0.0364) by CD4+CD25+FoxP3+ cells in the MLNs (figure 5D, E). The induction of AAD caused a statistically significant reduction in the proportion of FoxP3-expressing CD4+CD25+ cells, which was not observed in conjugate vaccine-treated mice (figure 5F).
We then hypothesised that as a result of the increased expression of CD103 and CTLA-4 and sustained expression of FoxP3, conjugate vaccine-induced Tregs would be more suppressive than those induced by OVA sensitisation and challenge alone. Indeed, CD4+CD25+ cells isolated from the MLNs of conjugate vaccine-treated mice suppressed CD4+CD25– OVA-induced effector T cell proliferation to a greater extent compared with untreated mice (1.5×103 CD4+CD25+ T cells from OVA-sensitised conjugate vaccine-treated mice suppressed CD4+CD25– effector T cells to an equivalent level to 12×103 CD4+CD25+ T cells from untreated OVA-sensitised mice; figure 5G). The suppression of OVA-induced T cell proliferation by Tregs also attenuated OVA-induced Th2 cytokine release (IL-5 and IL-13; not shown).
CD25 inactivation restores AAD following conjugate vaccine treatment
To confirm the role of conjugate vaccine-induced Tregs in suppressing AAD, Tregs were inactivated by anti-CD25 administration. Anti-CD25 administration restored AHR, eosinophil numbers in the BAL fluid and the levels of OVA-induced IL-5 and IL-13 released from MLN T cells to the levels observed in untreated OVA-sensitised and challenged mice (figure 6A–D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of anti-CD25 treatment on conjugate vaccine (CJ)-induced suppression of (A) airway hyper-responsiveness (AHR), (B) eosinophil numbers in bronchoalveolar lavage (BAL) and ovalbumin (OVA)-induced (C) interleukin 5 (IL-5) and (D) IL-13 release from mediastinal lymph node (MLN) T cells. Data represent the mean±SEM from six mice. Significant differences between saline-sensitised (Saline) and OVA-sensitised (OVA) controls are shown as #p<0.05, ##p<0.01. Significant differences between OVA-sensitised mice and anti-CD25- and CJ-treated OVA-sensitised mice are shown as *p<0.05, **p<0.01.
Discussion
Here we demonstrate that a S pneumoniae conjugate vaccine, which is currently licensed for human use, suppresses hallmark features of AAD in mouse models, indicating its potential as an immunoregulatory treatment for asthma. Treatment with the conjugate vaccine reduced Th2 cytokine release, cellular inflammation, antibody production and AHR. These inhibitory effects were mediated by increased numbers and/or percentages of local and systemic Tregs that had enhanced suppressive capacities.
The efficacy of the conjugate vaccine in preventing AAD compared with the lack of response to the polysaccharide vaccine may be attributed to their different composition. The conjugate vaccine is composed of seven types of pneumococcal polysaccharide that are conjugated to an immunogenic diphtheria toxoid, whereas the polysaccharide vaccine contains 23 serotypes of polysaccharide. The conjugate vaccine induces DC-driven T cell priming and elicits both T cell- and B cell-dependent antibody responses.17 In contrast, the polysaccharide vaccine induces T cell-independent antibody responses. Our results strongly suggest that to generate the induction of Tregs that suppress AAD, co-administration of polysaccharides with an immunogenic protein (conjugate vaccine) is required. Although polysaccharides alone (polysaccharide vaccine) are ineffective, the polysaccharide component of the conjugate vaccine is essential, since the backbone of the conjugate vaccine did not suppress AAD. The conjugate but not the polysaccharide vaccine contains AlPO4. It is unknown whether conjugate vaccine lacking AlPO4 would suppress AAD. However, ethanol-killed S pneumoniae, in the absence of AlPO4, effectively suppressed AAD.14 Therefore, it is likely that the AlPO4 component is not necessary and that the conjugate vaccine without AlPO4 would still be effective.
CpG-ODNs can prevent the induction of many features of AAD in mouse models and have been proposed as an immunoregulatory treatment for asthma. However, CpG-ODNs induce Th1 responses and are associated with increased IFNγ, which has proinflammatory activity in the lower airways that may promote AHR and increase the severity of AAD.18–21 In our study, CpG-ODNs induced T cell IFNγ release and not all features of AAD were attenuated. The conjugate vaccine was more effective at suppressing the features of AAD, and CpG-ODNs did not augment the effects of the conjugate vaccine. This is probably because CpG-ODNs promote Th1 responses whereas the conjugate vaccine is protective through the induction of Treg responses. CpG-ODN-induced IFNγ release was prevented by the conjugate vaccine, suggesting that the induction of Th1 responses by CpG-ODNs may be suppressed by conjugate vaccine-induced Tregs.
Importantly, intranasal administration of the conjugate vaccine was effective at suppressing established AAD. This provides support for the use of the conjugate vaccine in humans with established asthma. The conjugate vaccine may not offer instant relief during an asthma attack since the induction of Tregs is likely to take ∼3–4 days. Hence, the conjugate vaccine is likely to be most effective when used prophylactically. Intramuscular administration of the conjugate vaccine, which is the current route of administration to humans to prevent infection, had limited effects on AAD. This indicates that the vaccine must be delivered directly to the respiratory mucosa for greatest efficacy. We speculate that this may result from a requirement for the direct stimulation of DCs in the respiratory mucosa that subsequently enhances the suppression of AAD. It is possible that intranasal administration of the conjugate vaccine may protect against both asthma and infection, and this possibility will be investigated in future studies.
The conjugate vaccine promoted Treg expansion both during AAD and prior to OVA challenge. Tregs may be antigen specific or may have bystander effects that are independent of T cell receptor engagement.8 We speculate that conjugate vaccine-induced Tregs are not antigen specific since they are also able to suppress anti-CD3/anti-CD28-induced stimulation of effector T cells (not shown). If conjugate vaccine-induced Tregs are non-specific, they may predispose treated individuals to infection. However, subjects with asthma have reduced numbers and function of Tregs compared with those without asthma.3 Therefore, conjugate vaccine administration could be tailored to restore the balance of Tregs in subjects with asthma without excessive induction that would increase susceptibility to infection. Further research is required to fully elucidate the mechanisms of the Treg-mediated suppression of AAD in this study.
Suppression of Th2-mediated responses occurs both locally and systemically, since Th2 cytokine release in the MLNs and spleen, levels of circulating IgE and IgG1, and eosinophils in the lung and blood were attenuated. Our data show that the conjugate vaccine increased the total number of Tregs in the MLNs prior to challenge, and a higher proportion of CD4+ cells were CD25+FoxP3+ in the MLNs, lung and spleen after challenge. Hence, in our system it is likely that Treg expansion occurs in the MLNs and that OVA challenge promotes Treg trafficking and the prevention and suppression of inflammation at distal sites. This agrees with another study which showed that Tregs expand in the draining lymph nodes before moving to the inflammatory site in the skin.22 The extent of trafficking between the MLNs, lung and spleen in our study remains unknown. Lymph node Tregs express CCR7 and CCR6, whereas Tregs at the site of inflammation express CCR4 and CCR5, which may facilitate analytical tracking of Treg migration.23 Further investigation is required to understand conjugate vaccine-induced Treg trafficking and the chemokine receptors and ligands involved in the suppression of AAD.
A variety of Treg suppressive mechanisms of effector responses occur and are characterised by the nature of immune response, eliciting agent, site of inflammation, and the genetic and immunological background of the individual. IL-10 and TGFβ1 are immunosuppressive cytokines that may be released by Tregs to mediate suppression. Notably, IL-10 is a pleiotropic cytokine and is also released by Th2 cells in AAD. In our study, the conjugate vaccine suppressed release of IL-10 and TGFβ1 from MLN T cells, excluding their involvement in suppressing AAD. Our observations support previous investigations where a gastrointestinal nematode infestation suppressed AAD in mice. Adoptive transfer of nematode-induced Tregs from IL-10–/– mice transferred attenuation of Th2 responses and AAD.24 In addition, Schistosoma mansoni antigens suppress AAD independently of IL-10.25 Hence, IL-10 is not necessary for the attenuation of Th2 responses by Tregs. Others have also shown that suppression of effector T responses in vitro and in vivo does not require secretion of IL-10 or TGFβ1 by Tregs.24 26 27 Tregs may mediate suppression through cell contact-dependent mechanisms.
Notably, conjugate vaccine-induced Tregs were more suppressive on a per cell basis. CD25+ is also a marker of T cell activation and it is possible that CD4+CD25+ Tregs isolated from conjugate vaccine-treated mice had more Tregs and fewer effector T cells, and it was these changes that resulted in increased suppressive capacity. However, conjugate vaccine CD4+CD25+FoxP3+ cells had increased expression of the cell surface markers CD103 and CTLA-4, which are indicative of enhanced Treg functional capacity. CD103 is a marker of in vivo activated FoxP3+ Tregs, which have enhanced suppressive capacity.28 CTLA-4 expression inhibits CD80 and CD86 co-stimulation of T cells by DCs, and is required for FoxP3+ Treg function in vivo.29 Thus an increase in CTLA-4+FoxP3+ Tregs in conjugate vaccine-induced Tregs correlates with enhanced suppression of T cell proliferation. Subjects with asthma have reduced FoxP3 expression, and glucocorticoids may be effective, in part, by upregulating its expression.30 31 In our study, induction of AAD resulted in a reduced proportion of CD4+CD25+ cells expressing FoxP3; however, treatment with the conjugate vaccine maintained expression at preantigen exposure levels. Taken together, these results indicate that conjugate vaccine treatment leads to combined increases in CD103 and CTLA-4 and sustains the proportion of cells expressing FoxP3, resulting in a pool of Tregs with greater functional immunosuppressive capacity. Further studies are underway to elucidate other possible mechanisms of increased suppressive capacity of these Tregs.8
Administration of 100 μg of anti-CD25 depletes CD4+ cells that are CD25+ for up to 24 days.32 Furthermore, anti-CD25 has been employed to demonstrate an essential role for CD25+ Tregs in the suppression of AAD by S mansoni antigens.25 In our study, anti-CD25 administration inhibited the effects of the conjugate vaccine on AAD, supporting the concept that conjugate vaccine-induced CD25+ Tregs are essential for the suppression of disease. Notably, anti-CD25 may also deplete CD25+ effector T cells. However, when anti-CD25 was administered before the conjugate vaccine, complete restoration of AAD was achieved. This indicates that effector T cells were not affected and that Tregs were specifically depleted.
In addition to the suppression of effector cell responses by Tregs, the induction of Tregs may also result in deviation of T cell development away from the production of effector T cells. The expression of FoxP3 in naïve T cells fixes and stabilises their Treg lineage commitment, so that these cells cannot develop into effector T cells.33 The induction of Tregs may also prevent the establishment of Th17 cells and Th17-mediated disease. In the presence of TGFβ naïve T cells are induced to develop into Tregs, whereas the combination of TGFβ and IL-6 promotes Th17 cell differentiation. Treg-inducing agents may block or prevent the involvement of IL-6, thereby promoting Treg induction and preventing the development of Th17 cells. In AAD, blocking membrane-bound IL-6R, with anti-IL-6R antibody, induced Tregs and resulted in the attenuation of AAD.34 Therefore, in addition to the direct suppression of effector T cell responses, conjugate vaccine-induced Tregs may also suppress AAD via deviation of T cell development away from the production of effector T cells.
In summary, the S pneumoniae conjugate vaccine suppresses the critical features of AAD. The expansion of vaccine-induced Tregs that have a greater functional capacity underpins the mechanism of suppression of AAD. Utilisation of the conjugate vaccine as a Treg-inducing immunoregulatory treatment may provide a novel approach for the treatment of asthma.
References
Supplementary materials
Web Only Data thx.2009.131508
Files in this Data Supplement:
Footnotes
Funding The Hill family, Asthma Foundation NSW, CRC for Asthma and Airways, National Health and Medical Research Council (project grants 401238, 569219) and The University of Newcastle.
Competing interests None.
Ethics approval All experiments were approved by the Animal Care and Ethics Committee, University of Newcastle.
Provenance and peer review Not commissioned; externally peer reviewed.