Article Text

Statistics from Altmetric.com
S88 SOURCES OF INCREASED PLASMA SOLUBLE TNF RECEPTORS DURING INJURIOUS MECHANICAL VENTILATION IN MICE
A. D. Dorr, M. R. Wilson, K. P. O’Dea, M. Takata. Department of Anaesthetics, Pain Medicine and Intensive Care, Imperial College, London, UK
Introduction Increased plasma levels of soluble tumour necrosis factor (TNF) receptors (sTNFRs) are associated with mortality in patients with acute respiratory distress syndrome (ARDS) ventilated with high tidal volumes (VTs). These increases are considered to reflect either systemic inflammation, or decompartmentalisation of elevated intra-alveolar sTNFRs into the circulation due to alveolar–capillary barrier dysfunction. However, the contribution of mechanical ventilation per se has not been well defined. We have previously shown in an in vivo mouse model of pure ventilator-induced lung injury that sTNFRs can leak into the alveolar space from the plasma.1 Consequently, it is unclear where increases in plasma sTNFRs would originate.
Methods Anaesthetised C57BL6 mice were ventilated with high (36–41 ml/kg) or low VT (8–9 ml/kg) for up to 2 h. Upon termination, plasma samples were taken for quantification of sTNFR p55 and p75 (ELISA), and lungs were harvested for flow cytometric analysis of lung cell suspensions for TNFR p55 and p75 expression.
Results Plasma sTNFR p55 levels substantially increased at 1 h with high VT compared with low VT (see table 1), but declined at 2 h. A similar trend, though not statistically significant, was observed for sTNFR p75. After 2 h of high VT ventilation, pulmonary endothelial cells and lung-marginated monocytes had decreased surface expression of TNFRs. Lung-marginated neutrophils exhibited no changes in sTNFR expression.
Conclusions These data indicate that injurious ventilation can induce systemic sTNFR changes in the absence of pre-existing lung/systemic pathology. The increases in plasma sTNFRs occur earlier than previously reported sTNFR elevation in the alveolar space,1 and therefore cannot be explained by decompartmentalisation of elevated intra-alveolar sTNFRs. Conversely, our data strongly suggest that injurious lung stretch directly activates pulmonary intravascular cells (endothelial cells and lung-marginated monocytes), inducing shedding of cell surface TNFRs within the pulmonary microcirculation and release of these receptors into the systemic circulation. This could attenuate TNF signalling on these cells and modulate systemic TNF activity. These results may give new insights into how ventilation can modify TNF/TNFR signalling and propagate systemic inflammation leading to multiorgan failure, independently from the intra-alveolar milieu.
Funding: Funded by Westminster Medical School Research Trust and Wellcome Trust.
References
S89 EPITHELIAL TO MESENCHYMAL TRANSITION IN AN IN VITRO MODEL OF ACUTE RESPIRATORY DISTRESS SYNDROME
1C. M. O’Kane, 2V. d’Sousa, 2D. R. Thickett, 1D. F. McAuley. 1Respiratory Medicine Research Programme, Centre for Infection and Immunity, Queen’s University of Belfast, Belfast, UK, 2Lung Injury and Fibrosis Treatment Programme, School of Experimental and Clinical Medicine, University of Birmingham, Birmingham, UK
Background Fibroproliferation occurs early in the acute respiratory distress syndrome (ARDS), but resolves in the majority of survivors. Epithelial to mesenchymal transition (EMT) is a potentially reversible process by which the epithelium becomes a source of fibroblasts, losing the epithelial and gaining a mesenchymal phenotype, accompanied by the production of matrix-degrading enzymes (matrix metalloproteinase (MMP)-2/-9). A549 cells have been shown to undergo EMT upon treatment with transforming growth factor β (TGFβ) which is upregulated in ARDS. There are no data confirming that primary human alveolar epithelial cells undergo EMT in vitro. We hypothesised that EMT is a mechanism of the potentially reversible alveolar fibrosis that occurs in ARDS.
Methods Type II human alveolar epithelial (ATII) cells were isolated from normal lung tissue in 4 patients with normal lung function undergoing lobectomy. ATII and A549 cells were stimulated with TGFβ or bronchoalveolar lavage (BAL) fluid from patients with ARDS. At 72 h cell morphology was examined, and supernatants and lysates collected. Supernatants were analysed by zymography for MMP-2/-9. Lysates were probed by western blot for E-cadherin/Zo-1 (epithelial markers) and vimentin/α-smooth muscle actin (αSMA) (mesenchymal markers).
Results As with A549 cells ATII cells simulated with TGFβ developed a spindle-shaped morphology, losing tight cell–cell contact. TGFβ at 1 and 10 ng/ml caused loss of both intracellular E-cadherin and Zo-1 (fig 1a). TGFβ at 0.1 ng/ml was sufficient to induce αSMA and vimentin (fig 1a) in ATII cells. A549 cell lysates underwent similar changes in response to TGFβ (data not shown). ATII cells showed high basal MMP-9 secretion, with some upregulation in response to TGFβ (fig 1a). A549 cells in contrast produced minimal MMP-9: TGFβ upregulated MMP-2 >5-fold, p<0.05 in these cells. ARDS BAL induced morphological changes similar to TGFβ in A549 cells, and caused induction of vimentin (fig 1b), αSMA and MMP-2, with loss of Zo-1 (fig 1c).
Conclusion We confirm for the first time that human primary ATII cells can undergo EMT in vitro under the influence of TGFβ. In addition we have shown that the inflammatory milieu of the alveolus in ARDS can induce a fibroblast-like phenotype in A549 cells. Data suggest that EMT may contribute to fibrosis in lung injury.

ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; MMP-9, matrix metalloproteinase-9; αSMA, α-smooth muscle actin; TGFβ, transforming growth factor β.
S90 ROS-DEPENDENT ACTIVATION OF P38 MAPK IS REQUIRED FOR LPS-INDUCED UPREGULATION OF TNFα-CONVERTING ENZYME (TACE/ADAM-17) ACTIVITY ON PRIMARY HUMAN MONOCYTES
A. J. Scott, K. P. O’Dea, J. M. Handy, M. Takata. Department of Anaesthetics, Pain Medicine and Intensive Care, Imperial College London, London, UK
Introduction Tumour necrosis factor (TNF) has been demonstrated to play a key role in inflammatory disorders such as acute lung injury. We have shown previously that stimulation-induced increases in the activity of TNFα-converting enzyme (TACE/ADAM-17) on primary human monocytes are dependent upon the production of reactive oxygen species (ROS).1 While ROS were implicated, their mechanism of action was unknown. Reports in the literature suggest that ROS may activate p38 mitogen-activated protein kinase (MAPK). Here we investigated whether ROS-dependent activation of p38 MAPK could be responsible for lipopolysaccharide (LPS)-induced TACE activity upregulation.
Methods Primary human monocytes were isolated from peripheral blood and purified by negative immunomagnetic bead selection. Monocytes (85–90% purity) were stimulated with LPS in the presence or absence of antioxidants and selective inhibitors. TACE enzymatic activity was measured by a fluorescence resonance energy transfer peptide-based assay.2 Intracellular phospho-p38 expression and ROS production (inferred from dihydrorhodamine which fluoresces upon oxidation) were quantified by flow cytometry.
Results LPS simulation of primary human monocytes resulted in a rapid, membrane expression-independent increase in TACE activity (42±12 (SD) vs 116±31 fluorescence units (FU)/min for 105 monocytes, p<0.001), and an increase in intracellular ROS levels (1760±232 vs 2740±390 mean fluorescence intensity (MFI), p<0.001). These LPS-induced increases in TACE activity and ROS production were effectively abolished by the antioxidant N-acetyl-l-cysteine (NALC). LPS stimulation resulted in the activation of p38 MAPK, as demonstrated by increased expression of phospho-p38 (3±3 vs 25±6 MFI, p<0.001), which was also inhibited by NALC (25±6 vs 14±4 MFI, p<0.05). An inhibitor of p38 MAPK, SB203580, substantially attenuated LPS-induced TACE activity upregulation (93±12 vs 27±9 FU/min, p<0.001).
Conclusion These results demonstrate that LPS-induced TACE activity upregulation depends upon two distinct signalling elements: first, production of ROS and secondly ROS-dependent activation of p38 MAPK. Inhibition of either p38 activity or ROS production can attenuate TACE activity upregulation. The findings suggest that the ROS–p38 axis could be exploited to reduce upregulation of TACE activity and consequent TNF shedding/release in inflammatory conditions such as acute lung injury.
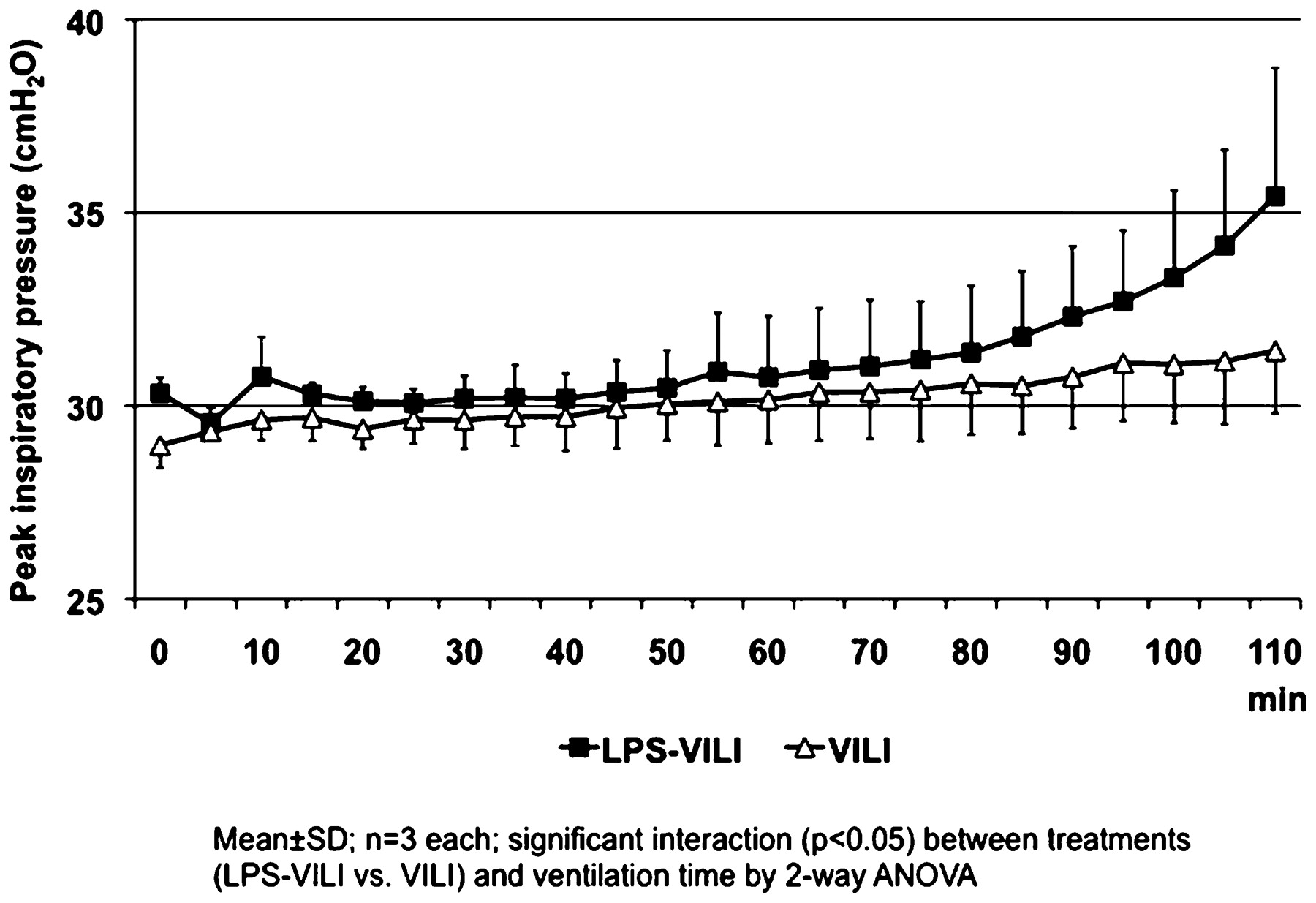
S91 ROLE OF INTRAVASCULAR LEUCOCYTES IN VENTILATOR-INDUCED LUNG INJURY IN THE ISOLATED PERFUSED MOUSE LUNG
K. Wakabayashi, M. R. Wilson, K. P. O’Dea, M. Takata. Department of Anaesthetics, Pain Medicine and Intensive Care, Imperial College London, London, UK
Introduction Recruitment of leucocytes to the lung and their interaction with pulmonary endothelium are considered to play important roles in the pathophysiology of acute lung injury. We have recently shown in an in vivo mouse model of ventilator-induced lung injury (VILI) that subclinical endotoxaemia sensitises the lung toward VILI, and monocytes marginated to the pulmonary microvasculature appear to play a key role in this phenomenon.1 However, it is intrinsically difficult in such in vivo models to differentiate clearly the effects of lung-marginated leucocytes from those of circulating leucocytes. Here we attempt to clarify the role of lung-marginated leucocytes in VILI using an ex vivo mouse isolated perfused lung (IPL) model.
Methods Lungs were obtained from C57BL6 mice, either untreated or pretreated in vivo with a subclinical dose of lipopolysaccharide (LPS; 20 ng ivintravenously, 2 h before lung perfusion). The lungs were perfused (25 ml/kg/min) with non-blood buffer in a recirculating manner and ventilated with high tidal volume (25–30 ml/kg, positive end-expiratory pressure (PEEP) 3 cm H2O) for 2 h. Lung lavage was performed at the end of experiments to determine the degree of pulmonary oedema.
Results LPS challenge promoted margination of large numbers of neutrophils (1.71±0.34 vs 0.13±0.03×106 cells/lung; p<0.01) and Gr-1high inflammatory subset monocytes (1.22±0.17 vs 0.21±0.04×106 cells/lung; p<0.001) to the lungs, as measured by flow cytometric analysis of lung cell suspensions. LPS pretreatment substantially exacerbated the development of VILI in this IPL model, as represented by more rapid increases in peak inspiratory pressure (see fig 1, p<0.05) and higher levels of total protein in lung lavage fluid (3.99±0.40 vs 1.48±0.25 mg/ml; p<0.001).
Conclusions These results confirm previous findings by ourselves and others that subclinical endotoxaemia sensitises the lung to the consequences of injurious mechanical ventilation. Importantly, our ex vivo IPL model demonstrated that this sensitisation can occur in the absence of circulating blood, adding further support to the critical role of “lung-marginated” leucocytes in the progression of VILI. The molecular mechanisms underlying this sensitisation remain to be elucidated, and the IPL will be a useful tool in these investigations.
Funding: This work was supported by the Wellcome Trust and BBSRC.
{kind=link}
{kind=link}
ANOVA, analysis of variance; LPS, lipopolysaccharide; VILI, ventilator-induced lung injury.
References
S92 DIFFERENTIAL ACTIVATION OF THE P38 MAP KINASE PATHWAY IN MOUSE MONOCYTE SUBSETS DURING CO-CULTURE WITH PULMONARY ENDOTHELIAL CELLS
J. O. Dokpesi, K. P. O’Dea, M. Takata. Department of Anaesthetics, Pain Medicine and Intensive Care, Imperial College London, UK
Introduction We recently demonstrated that lung-marginated “inflammatory” Gr-1high monocytes play a significant role in mouse models of sepsis-associated and ventilator-induced lung injury.1 2 The Gr-1high subset expresses higher levels of tumour necrosis factor α (TNF) than the “resident” Gr-1low subset in response to lipopolysaccharide (LPS), both within the lung microvasculature in vivo1 and in co-culture with primary mouse lung endothelial cells (MLECs) in vitro.5 Here, we investigated the role of the p38 mitogen-activated protein kinase (MAPK) pathway, an important regulator of TNF expression and other key inflammatory mediators, in determining monocyte subset TNF production during co-culture with MLECs.
Methods MLECs were isolated from C57BL6 mice. Peripheral blood mononuclear cells (PBMCs) from the same mouse strain were added to confluent MLEC monolayers and then stimulated with 100 ng/ml LPS. Levels of phosphorylated p38 and its downstream target, MK-2, were quantified in permeabilised/fixed monocytes by flow cytometry using phosphokinase-specific antibodies. The contribution of the p38 pathway to TNF expression was assessed using the p38 inhibitor SB203580.
Results At 15 min after LPS treatment, levels of phosphorylated p38 and MK2 increased markedly from baseline in Gr-1high monocytes (p38, 44.9±1.3 (SD) vs 20.9±1.3 mean fluorescence intensity (MFI); MK-2, 75.9±3.0 vs 10.3±1.0 MFI). In Gr-1low monocytes, phosphorylation of both kinases increased (p38, 22.5±1.3 vs 17.7±1.8 MFI; MK-2, 36.6±1.7 vs 17.0±0.8 MFI). but the levels were substantially lower than in Gr-1high monocytes (p<0.001). SB203580 completely inhibited LPS-induced expression of membrane TNF in both Gr-1high and Gr-1low monocytes.
Conclusion These results strongly suggest that the p38 MAPK pathway is a critical determinant of the differential TNF response exhibited by lung-marginated monocyte subsets during endotoxaemia in mice. The p38 MAPK pathway is therefore likely to play a pivotal role in monocyte-induced pulmonary inflammation during sepsis-associated acute lung injury, determining the expression levels of TNF and the other inflammatory genes it regulates.
Funding: This work was supported in part by BBSRC.
S93 INFLUENCE OF CARDIAC SURGERY UTILISING CARDIOPULMONARY BYPASS ON NEUTROPHIL-RELATED PROINFLAMMATORY RESPONSES
E. F. Goode, L. R. Hector, A. L. Lagan, D. D. Melley, G. J. Quinlan, R. E. Bundy. Department of Critical Care Medicine, National Heart and Lung Institute, Royal Brompton Hospital NHS Foundation Trust, Imperial College, London, UK
Introduction The systemic inflammatory response syndrome and its serious respiratory manifestations, including acute lung injury (ALI), are complications of cardiac surgery using cardiopulmonary bypass (CPB). Cardiac surgery is associated with release of inflammatory mediators. CPB may contribute to this response, and off-pump surgery may be associated with reduced levels of postoperative inflammation.
Neutrophils are crucial in the inflammatory response, releasing many proinflammatory mediators. Spontaneous neutrophil apoptosis is essential for the resolution of inflammation in ALI. We have previously shown that CPB delays neutrophil apoptosis. We hypothesised that neutrophil apoptosis is regulated by haem oxygenase-1 (HO-1) through haemoglobin-dependent mechanisms.
Objectives First, to investigate the relationship between haemolysis, induction of HO-1 and neutrophil apoptosis during cardiac surgery with CPB and without CPB (OPCAB). Secondly, to explore the influence of haem-containing modulators of the HO-1 pathway on viability and apoptosis of neutrophils obtained from healthy volunteers.
Methods Nine patients undergoing coronary artery bypass grafting surgery were recruited (5 CPB, 4 OPCAB). Plasma levels of free haemoglobin (Hb) were analysed. The expression of HO-1 was studied by RT-PCR. Neutrophil apoptosis was detected in blood collected preoperatively or 2 h following CPB and OPCAB by flow cytometry and morphological assessment following neutrophil isolation and incubation for 20 h.
Results Hb levels were 0.26 ±0.12 g/l post-CPB, but no free Hb was detected in OPCAB patients. HO-1 mRNA expression showed 0.91±0.29 copies/μl pre-CPB vs 1.56±0.65 copies/μl post-CPB, whereas OPCAB levels were 0.85±0.23 copies/μl presurgery vs 0.57±0.26 copies/μl postsurgery. Neutrophil viability following 20 h culture was 32.85±4.11% for control presurgery and rose 1.74±0.45% 2 h post-CPB vs 1.27±0.14% post-OPCAB. HO-1 activity inhibition by SnPP (tin protoporphyrin) after 20 h reduced neutrophil viability dose dependently (47.99±8.1% control, 45.05±4.32% 40 μM SnPP, 38.79±2.32% 80 μM SnPP). Treatment of volunteer neutrophils with the HO-1 inducer haem showed cell viability 172.2% of control, which was partially reversed by SnPP 40 μM (138.9±52.1%).
Conclusion Surgery increased neutrophil lifespan with a seemingly greater effect following CPB. Haemolysis during CPB and an associated induction of HO-1 may promote neutrophil viability and contribute to a prolonged inflammatory response, thereby potentially increasing the risk of ALI.