Article Text

Abstract
Background: In patients with symptoms suggestive of cystic fibrosis (CF) and intermediate sweat chloride values (30–60 mmol/l), extensive CFTR gene mutation analysis and nasal potential difference (NPD) measurement are used as additional diagnostic tests and a positive result in either test provides evidence of CFTR dysfunction. To define the phenotype of such patients and confirm the validity of grouping them, patients with intermediate sweat chloride values in whom either additional CF diagnostic test was abnormal were compared with subjects in whom this was not the case and patients with classic CF.
Methods: The phenotypic features of four groups were compared: 59 patients with CFTR dysfunction, 46 with an intermediate sweat chloride concentration but no evidence of CFTR dysfunction (CF unlikely), 103 patients with CF and pancreatic sufficiency (CF-PS) and 62 with CF and pancreatic insufficiency (CF-PI).
Results: The CFTR dysfunction group had more lower respiratory tract infections (p = 0.01), more isolation of CF pathogens (p<0.001) and clubbing (p = 0.001) than the CF unlikely group, but less frequent respiratory tract infections with CF pathogens than the CF-PS group (p = 0.05). Patients in the CF-PS group had a milder phenotype than those with PI. Many features showed stepwise changes through the patient groups.
Conclusion: Patients with intermediate sweat chloride values and two CFTR mutations or an abnormal NPD measurement have a CF-like phenotype compatible with CFTR dysfunction and, as a group, differ phenotypically from patients with intermediate sweat chloride values in whom further CF diagnostic tests are normal as well as from CF-PS and CF-PI patients.
Statistics from Altmetric.com
The diagnosis of cystic fibrosis (CF) is obvious when patients present in the first months of life with symptoms such as steatorrhoea and chronic cough. This clinical diagnosis is then confirmed by a sweat chloride value >60 mmol/l.1 2 However, since the discovery of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, it has become apparent that CF is not always a clearcut diagnosis; there is a wide clinical spectrum of diseases associated with CFTR mutations.3–5 Patients carrying two CFTR mutations can have milder symptoms that only become apparent during adolescence or adulthood, or can even present with a single clinical problem such as male infertility or pancreatitis.5 6 In patients with this milder phenotype, the sweat chloride concentration is often in the intermediate range (ie, 30–60 mmol/l).7 8 Diagnosis of CF is then supported by CFTR mutation analysis and/or bioassays of CFTR protein function such as nasal potential difference (NPD) measurement.1 9
In the algorithms proposed by the European Consensus Group,9 patients with symptoms suggestive of CF are classified according to the result of the pilocarpine sweat test: chloride >60 mmol/l, 30–60 mmol/l and <30 mmol/l (fig 1). Patients with appropriate symptoms and a sweat chloride level >60 mmol/l are considered as having CF since a sweat chloride level >60 mmol/l is almost always associated with CF.2 They may either have pancreatic insufficiency (CF-PI) or pancreatic sufficiency (CF-PS). In patients with a sweat chloride level <30 mmol/l, the diagnosis of CF is unlikely and an alternative diagnosis should be considered. In patients with symptoms suggestive of CF and a sweat chloride level in the intermediate range (30–60 mmol/l), further diagnostic tests should be done: extended search for CFTR mutations and/or NPD measurement. A positive result of either test provides evidence of CFTR dysfunction and thus a diagnosis of atypical or non-classic CF.

European diagnostic algorithm (reproduced with permission from De Boeck et al9). CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; FU, follow up; PD, potential difference.
The aim of the current study was to further define the disease phenotype of patients with intermediate sweat chloride values and evidence of CFTR dysfunction (by two CFTR mutations identified or an abnormal NPD result), as well as to justify grouping these patients as a separate phenotype in the European diagnostic consensus. The phenotype of patients with intermediate sweat chloride values and evidence of CFTR dysfunction was therefore compared with the phenotype of subjects with intermediate sweat chloride values but no further evidence of CFTR dysfunction. In addition, the phenotype of patients with CFTR dysfunction was compared with that of patients with a sweat chloride level >60 mmol/l and PS or PI.
METHODS
Source of data
Physicians at CF centres were approached through the European Coordination Action for Research in Cystic Fibrosis (EurocareCF, http://www.eurocarecf.eu) and the European Cystic Fibrosis Society (ECFS) Diagnostic Network Working Group (http://www.ecfsoc.org). A standardised data collection tool was developed and physicians completed these for all patients in their clinic belonging to one of the following groups:
Subjects with CF, a sweat chloride value >60 mmol/l and PS (CF PS).
Subjects with a sweat chloride value in the intermediate range (30–60 mmol/l) plus two CFTR mutations detected or an abnormal NPD measurement (termed CFTR dysfunction).
Subjects with a sweat chloride concentration in the intermediate range (30–60 mmol/l) but no or one CFTR mutation detected after extensive sequencing and/or a normal NPD measurement (termed CF unlikely).
For comparison, a group of CF patients homozygous for the F508del mutation was also randomly selected from the CF patient database of the University Hospital of Leuven. Group definitions are listed in table 1.
Since the aim was to compare disease presentation and severity in different groups, patients identified through newborn screening and patients who had undergone lung transplantation were excluded from analysis.
Diagnostic tests
Only sweat chloride concentrations determined according to current guidelines by the Gibson and Cooke method (pilocarpine iontophoresis) were recorded.2 If patients had multiple measurements, the mean sweat chloride concentration was reported. As in the European consensus,9 the intermediate sweat test chloride range was defined as 30–60 mmol/l because finding two CFTR mutations in patients with CF-like symptoms is equally frequent in patients with sweat chloride values of 30–40 mmol/l and 40–60 mmol/l.8
For further data analysis, CFTR mutations detected were grouped according to mutation class.10–13 For rare mutations a mutation class could not be reliably assigned. Classes I, II and III are more often associated with “severe” disease than mutation classes IV and V.
NPD measurements were performed according to the standard protocols.14 15 A change after zero chloride plus isoproterenol smaller than −5 mV or a ratio of total chloride response to amiloride response of >0.35 were considered abnormal.16 The validity of NPD measurements has recently been demonstrated with studies showing long-term repeatability and consistency.17 18
Clinical characteristics
Age at diagnosis was derived from the date of the first sweat test performed. Exocrine pancreatic sufficiency (PS) was defined as faecal fat loss <7 g/day, fat absorption >93% or a fecal elastase level of >200 μg/g faeces. Weight, height and body mass index (BMI) at most recent clinical evaluation were expressed as Z-scores.19 Lung function tests were performed according to the American Thoracic Society standards and forced expiratory volume in 1 s (FEV1) was expressed as percentage predicted (FEV1% pred).20 21 Chronic Pseudomonas aeruginosa infection was reported using the European consensus definition.22 The occurrence of recurrent lower respiratory tract infections (LRTI), regular expectoration of sputum, isolation of P aeruginosa or Staphylococcus aureus from respiratory cultures ever, clubbing and nasal polyposis were graded as absent or present. Allergic bronchopulmonary aspergillosis (ABPA) was defined following the American consensus criteria.23
Pancreatitis was defined as an episode of acute abdominal pain associated with serum amylase levels more than 1.5 times above the upper limit of normal established by the individual laboratories.24 The absence or presence of meconium ileus (MI) at birth was registered. Distal intestinal obstruction syndrome (DIOS) was defined using a recent consensus definition.25 For CF liver disease the definition by Colombo26 was used and for portal hypertension (PHT) the definition by Sokol27 was used.
Analysis of data
For continuous variables with a normal distribution, initial ANOVA was performed. Post hoc analysis was performed using Tukey if equal variance could be assumed and using Tamhane’s otherwise. For continuous variables without normal distribution the Kruskal-Wallis test and Mann-Whitney U test were used. Continuous variables with normal distribution were expressed as means and standard deviation (SD); otherwise medians and interquartile range are reported. Categorical data were compared with the Pearson χ2 test (or the Fisher exact test for numbers below 5). Analyses were performed using SPSS Version 15.0; p values of ⩽0.05 were considered statistically significant.
RESULTS
Data inclusion, criteria used for group assignment, CFTR mutations detected in the patient groups
Thirteen centres from 10 countries contributed data on 401 patients. Data were excluded on 131 patients, 34 because the sweat chloride result was missing, <30 mmol/l or in the intermediate range (30–60 mmol/l) but no further evaluation of CFTR dysfunction by mutation analysis nor NPD; 65 with key data missing (date of birth, date of clinical assessment, weight, height and lung function data); and 32 patients with CF diagnosed from newborn screening programmes. Data from 270 patients were thus included in the comparison of the four groups: CFTR dysfunction (n = 59), CF unlikely (n = 46), CF-PS (n = 103) and CF-PI (n = 62).
Of the 59 patients with intermediate sweat chloride values and CFTR dysfunction, 31 (52%) had two CFTR mutations identified, 14 (24%) were diagnosed by abnormal NPD and 14 (24%) had two CFTR mutations identified as well as an abnormal NPD. Of the 46 patients with intermediate sweat chloride values classified as “CF unlikely”, 27 (59%) were because they had no or one mutation identified after mutation screening plus a normal NPD, 18 (39%) because they had no or only one mutation detected after CFTR gene sequencing, 1 (2%) because they had two mutations detected with sequencing (one being silent: ie, E528E) but a normal NPD.
The list of mutations per patient group is given in table 2 and the distribution of CFTR mutation classes in table 3. Many patients with CFTR dysfunction (58%) had one class I–III plus one class IV–V mutation. Most patients (87%) in the “CF unlikely” group did not have any mutation identified; four patients (9%) were F508del carriers and one patient carried the IVS8-5T. In one subject two mutations were detected, one of which was a silent mutation (E528E), and this subject had a normal NPD result. Three patients with CF-PS and two patients with CFTR dysfunction carried two class I–III mutations. All five had the F508del mutation associated with S945L, a mutation considered as a class II mutation.
Comparison of the four patient groups
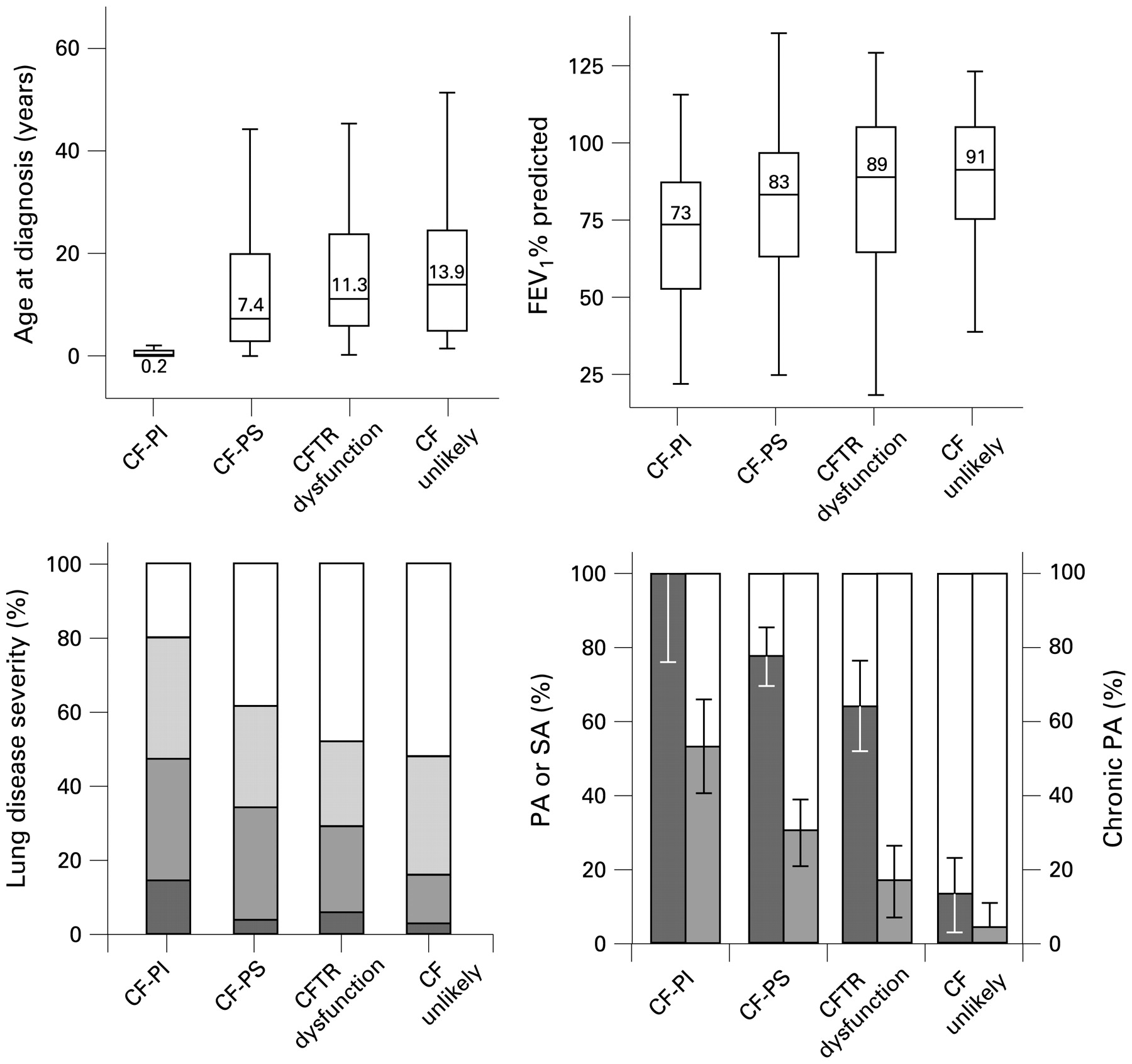
The median age was 17–20 years and did not differ between the patient groups. Full results are shown in table 4 and fig 2. The major differences are reported below.

{kind=link}
{kind=link}
For the patient groups CF-PI, CF-PS, CFTR dysfunction and CF unlikely, the following are shown: (A) Box plots of age at diagnosis (numbers in boxes represent median values). (B) Box plots of FEV1% predicted (numbers in boxes represent median values. (C) Distribution of lung disease severity according to FEV1% predicted (normal ⩾90% (white), mild 70–89% (pale), moderate 40–69% (grey), severe <40% (dark)). (D) Percentage of patients with isolation of Pseudomonas aeruginosa (PA) or Staphylococcal aureus (SA) from the respiratory tract (dark); percentage of subjects with chronic P aeruginosa colonisation (grey). Error bars represent 95% confidence intervals. CF-PI, sweat chloride concentration >60 mmol/l and pancreatic insufficiency; CF-PS, sweat chloride concentration >60 mmol/l and pancreatic sufficiency; CFTR dysfunction, sweat chloride concentration 30–60 mmol/l and two mutations identified and/or abnormal nasal potential difference (NPD); CF unlikely, patients with an intermediate sweat chloride concentration but no detection of two CFTR mutations and/or normal NPD; FEV1, forced expiratory volume in 1 s.
CFTR dysfunction versus CF unlikely
A larger proportion of patients with CFTR dysfunction had clubbing (p = 0.001), recurrent LRTI (p = 0.01) and positive isolation of P aeruginosa or S aureus (p<0.001). Patients with CFTR dysfunction tended to have more frequent chronic P aeruginosa colonisation (17% vs 5%; p = 0.07).
CFTR dysfunction versus CF-PS
Patients with CFTR dysfunction were diagnosed at an older age (p = 0.03), had less frequently positive isolates of P aeruginosa or S aureus (p = 0.05) and a trend to less chronic P aeruginosa colonisation (17% vs 30%; p = 0.06) and higher median FEV1% pred (89% vs 83%; p = 0.15). The bowel complications were also differentiating, with MI and/or DIOS (p = 0.05) and liver disease (p = 0.04) less frequent in patients with CFTR dysfunction than in patients CF-PS.
CF-PI versus CF-PS
Patients with CF-PI had a more severe phenotype than patients with CF-PS for most variables evaluated, except pancreatitis that was more frequent in patients with CF-PS (14% vs 2%; p = 0.01).
DISCUSSION
We have retrospectively collected phenotypic features of patients with intermediate sweat chloride values (30–60 mmol/l) and CFTR dysfunction defined as detection of two CFTR mutations and/or an abnormal NPD and compared them with the phenotypic features of patients with intermediate sweat chloride values but no abnormalities in further CF diagnostic tests who are thus unlikely to suffer from CF. Patients in the group with CFTR dysfunction more often exhibit recurrent lung infections and more often carry CF pathogens than patients in the “CF unlikely” group. Patients with CFTR dysfunction also differ significantly from patients with CF-PS; they are less likely to have P aeruginosa and/or S aureus infection and their median age at diagnosis is higher, reflecting the fact that they present with symptoms later and/or that these symptoms are less suggestive of a possible CF phenotype. MI/DIOS and liver disease are also less frequent in patients with CFTR dysfunction than in those with CF-PS. Overall, the current data therefore support the concept that, as a group, patients with intermediate sweat chloride values and CFTR dysfunction have an intermediate phenotype between patients with CF-PS and subjects in whom CF is unlikely. Delineating them as a separate group called “atypical or non-classic CF” as has been done in the European algorithm is thus justified, even if there is some overlap between the groups.9
Making a distinction between disease groups is helpful in discussions with patients. The knowledge that, on the whole, patients with an intermediate sweat chloride value and CFTR dysfunction are more likely to have a milder disease phenotype can be of some comfort for these patients. The fact that, in many of these patients, the diagnosis is made at a later age is concordant with this milder disease expression. Follow-up remains important since disease manifestations may change over time, and also patients with sweat chloride values 30–60 mmol/l and CFTR dysfunction are at risk of developing serious disease, especially P aeruginosa lung infection.28 It may be extrapolated that timely treatment and other healthcare measures will benefit patients with CFTR dysfunction just as has been proved for patients with CF.29 In the current study, differences between patients with CFTR dysfunction and those in the “CF unlikely” group were mainly found for the lung phenotype. This has been reported by other groups.30 To differentiate patients with what these authors called “atypical CF” from patients not meeting CF diagnostic criteria, a history of isolation of P aeruginosa from respiratory specimens was considered more useful than the presence of steatorrhoea.
Parameters other than sweat test results have been used to group or classify patients, including disease manifestations, CFTR gene mutation analysis and NPD results. All have their advantages and disadvantages. No single parameter is likely to be ideal, since we know that disease severity depends not only on the CFTR mutation class31 but also on genetic modifiers32 33 and environmental factors such as optimal prevention and treatment of pulmonary exacerbations.34 35
Some use the clinical picture to describe patient groups and use the terms “atypical CF”, “non-classic CF” and “CFTR-opathy” to describe patients with disease manifestations that are very mild or only occur in one organ.36 37 Just using disease manifestations to classify patients is inaccurate, since age and intensity of treatment certainly influence the disease course and can thus even obscure the true phenotypic differences between the groups. We agree, however, that considering the patient’s symptoms is very important and feel that it is impossible to have just one diagnostic algorithm that is appropriate for subjects identified after newborn screening as well as for subjects with a history of symptoms suggestive of CF. All consensus statements on the diagnosis of CF—including the revised American consensus—state that the clinical picture is very important in making a diagnosis of CF. We thus agree with the consensus document on CFTR mutations that the mere identification of two CFTR mutations is not sufficient to make a diagnosis of CF or of CFTR dysfunction,13 which is why extreme caution should be used in the case of newborn screening. If, however, a subject has a (longstanding) history of symptoms that are compatible with CF (the mean age of the patients in the report is 18–20 years) plus an intermediate sweat test value, and if in such an individual two CFTR mutations are being identified, these combined findings are sufficient evidence for a diagnosis of CFTR dysfunction.
In the revised American diagnostic consensus, CFTR mutation analysis is, to a large degree, used to classify patients as suffering from CF, CFTR-related disorder or CF very unlikely.38 Patients are classified as having CF if they have a sweat chloride value ⩾60 mmol/l or if they have two CF-causing mutations. In patients with intermediate sweat chloride values, extensive CFTR mutation analysis is advised. Extreme caution should be used, however, in genotype interpretation; only 28 mutations are considered as causing CF and a limited list of mutations is proposed as being “associated with CFTR-related disorders”.13 These lists are difficult to use because nine mutations are classified as both CF-causing and being associated with CFTR-related disorders. The current data set also demonstrates the difficulty of using mutation analysis to describe disease severity. Indeed, carrying at least one class IV–V mutation was more common in patients with intermediate sweat chloride levels and CFTR dysfunction than in patients with CF-PS, but there is significant overlap in mutation distribution between patient groups. This is in agreement with the importance of gene modifiers and environmental factors.
Determining the functional consequences of a CFTR mutation and distinguishing a CF-causing mutation from a silent sequence variant is challenging and would require another “gold standard” test to document the absolute presence of disease. This is where the value of NPD should be considered. NPD was indeed considered a valid diagnostic test in the initial American CF consensus guideline1 but has been reduced to the status of “only providing contributory evidence in the diagnostic evaluation” in the revised consensus.38 The authors stress the absence of generally accepted cut-off values as well as the limited availability of the test. We argue that NPD has a long track record of aiding in CF diagnosis,39 is useful to rule out CFTR dysfunction16 and the test result is consistent over time.18 Although there are no absolute criteria for what constitutes an abnormal NPD result, the ratio of chloride to amiloride response provides the best distinction between CF and normal.16–18 In the current data set, most patients (19/28) in the CFTR dysfunction group had an abnormal chloride response as well as an abnormal chloride to amiloride response ratio, but in some patients (9/28) the results were discordant. In the present cohort many patients had CFTR analysis as well as NPD and the test results concurred; patients with two CFTR mutations identified had an abnormal NPD and patients with only one mutation identified had a normal NPD. There was one exception—one patient had two CFTR mutations, one of which was silent (E528E); this patient had a normal NPD, further supporting the value of this test. We would argue that mutation analysis is—although commercially available—equally fraught with problems since genotype data interpretation is so complex and, in many situations, finding two CFTR mutations does not settle the diagnostic issue. Even if, in the current data collection, the selection of patients with proven CFTR dysfunction is restricted to patients with both NPD parameters abnormal and only accepting mutation R117H or 5T when associated with TG13 or TG12 (n = 45), the study conclusions that patients with CFTR dysfunction differ from patients with “CF unlikely” remain steadfast (see online supplement).
There is no consensus at present as to the most appropriate terminology for patients with intermediate sweat chloride values and two CFTR mutations identified. The most recent American consensus uses the term “CFTR-related disorder”.38 This term is then used in a broader context than before (ie, to refer to patients with two CFTR mutations identified); many of these patients would previously have been described as “atypical CF”. When applying the recently published “mutation guidelines”13 to our patient cohort with intermediate sweat chloride values, 12/59 patients would be called “CF-PS” and not “CFTR dysfunction” (ie, patients carrying 3849+10 kb C>T plus F508del (n = 11) or R1162X (n = 1)). At least eight additional patients with CFTR dysfunction would be classified as suffering from either CF or a CFTR-related disorder. In the cohorts described in this study, the CFTR mutations are identified in patients with disease manifestations compatible with CF, not in subjects identified by newborn screening. For most if not all mutations identified in the current cohort, the CFTR mutation database10 contains patient reports with clear CF disease manifestations. Most patients with two CFTR mutations identified are likely to be at risk of developing severe lung disease, and the current data collection again supports this statement, the occurrence of CF lung pathogens being more frequent in patients with CFTR dysfunction than in those in the “CF unlikely” group. The current classification is not ideal since our knowledge about the long-term consequence of many CFTR mutations is insufficient. Ideas will therefore continue to evolve. Changing disease terminology may, however, not lead to excluding patients with CFTR dysfunction or CFTR-related disorders from access to specific drugs. Also, in this respect, our group at present prefers the term “atypical or non-classic CF” for patients with CFTR dysfunction.
There is no consensus on what lower limit to use as the sweat chloride cut-off value. American guidelines2 still recommend that sweat chloride concentrations <40 mmol/l are considered as normal; only in infants is the lower cut-off value of 30 mmol/l used. We have previously argued that 30 mmol/l is a better cut-off value to use when investigating subjects with CF-like symptoms.8 9 In the present study, 16/59 patients (27%) with an intermediate sweat test value and proven CFTR dysfunction had a concentration in the range 30–40 mmol/l. There was no difference in the median sweat chloride concentration between subjects with and without CFTR dysfunction. In patients with symptoms compatible with a clinical diagnosis of CFTR dysfunction, using 30 mmol/l as a cut-off value will decrease the number of false negatives. Thus, we reaffirm our previous lower limit of >30 mmol/l.
Taking all these factors into account, we argue that the sweat test result still provides a useful distinction between patient groups. The sweat test is a key diagnostic test for CF and has proved remarkably robust. The sweat test result is closely linked to the basic pathophysiology of CF. In cross-sectional studies of patients with various CFTR mutations, it has been shown repeatedly that mean sweat chloride values differ between patient groups. In a recent study by Wilschanski et al,40 the sweat chloride result discriminated between patient groups better than the results from NPD measurements. Rowe et al41 made the point that sweat chloride values are a marker of disease severity; they plotted sweat chloride concentrations against percentage CFTR activity and discussed the potential use of sweat chloride results as an outcome variable in CF trials. In addition to showing group differences, the distinction between CF and CFTR dysfunction on the basis of sweat chloride values, as used in the European consensus,9 is straightforward and easy to apply. In patients with classic CF the diagnosis is clearcut; they have CF-associated symptoms and a positive sweat test. The group with non-classic or atypical CF are those with an intermediate sweat chloride value in whom CFTR dysfunction is proved by the additional diagnostic tests (ie, extensive CFTR gene analysis or abnormal NPD measurement).
Given the overlap in phenotypic features, sweat test results, mutation analysis and in NPD results, some authors prefer the concept that the clinical manifestations of CF form a continuum.11 The gradual change in phenotype in the current study (fig 2) is consistent with both the continuum and the stepwise arguments.
In conclusion, when applying the European Consensus Diagnostic Group definitions and dividing the patients into CF and CFTR dysfunction on the basis of sweat chloride values, we have shown that, as a group, patients with CF have more severe disease than patients with CFTR dysfunction. Furthermore, patients with CFTR dysfunction have a more severe pulmonary phenotype than subjects with an intermediate sweat chloride concentration in whom CF is considered unlikely. Making a distinction between patients with CF and those with CFTR dysfunction therefore seems appropriate.
Acknowledgments
The authors thank Harry Cuppens for advise on genotypes, Yasmin Yaakov and Francois Vermeulen for help in data collection, C Castellani and B Assael from the Verona CF centre for data contribution and Els Aertgeerts for secretarial assistance.
REFERENCES
Supplementary materials
Web only appendix 64:8;683
Files in this Data Supplement:
Footnotes
Competing interests: None.
Funding: EuroCareCF was supported by the European Community’s Sixth Framework; Programme for Research, Priority 1 “Life Sciences, Genomics and Biotechnology for Health”, contract number LSHM-CT-2005-018932.
Ethics approval: The study was approved by the ethics committee of the University Hospital of Leuven.
▸ Additional data are published online only at http://thorax.bmj.com/content/vol64/issue8