Article Text

Abstract
Background: Smoking increases the susceptibility to pulmonary infection and is a risk factor for the development of chronic obstructive pulmonary disease (COPD). It is postulated that cigarette smoke suppresses the activation of the innate immune system in response to bacterial infection.
Methods: Using sensitive ex vivo analysis, the level of the endogenous antibiotic peptide human β-defensin-2 (hBD-2) was measured in pharyngeal washing fluid and sputum from patients with community acquired pneumonia. The regulation of antibacterial host defence molecules was studied in vitro. The effect of cigarette smoke on the antibacterial activity of differentiated airway epithelium and the expression of host defence molecules was studied in an in vitro infection model.
Results: Current or former smoking was associated with significantly reduced hBD-2 levels in pharyngeal washing fluid and sputum from patients with acute pneumonia. Exposure of airway epithelium to smoke in vitro inhibited the induction of hBD-2 by bacteria. This correlated with decreased antimicrobial activity. This effect was mimicked by hydrogen peroxide, and catalase blunted the smoke-induced inhibition of epithelial host defence.
Conclusions: Smoke exposure suppresses the induction of epithelial antibacterial host defences. These findings link smoking with increased susceptibility to infection. This mechanism may be important in the pathogenesis of pneumonia and COPD.
Statistics from Altmetric.com
Cigarette smoking and exposure to environmental tobacco smoke increase the risk of pulmonary infections in general and the risk to contract invasive pneumococcal disease by a factor of 4.1.1 In addition, other pulmonary infections are more frequent in smokers including influenza and tuberculosis.2 A population-based case-control study revealed that the risk of community acquired pneumonia (CAP) attributable to the consumption of any type of tobacco was 32.4% in patients with chronic obstructive pulmonary disease (COPD).3 Animal experiments have shown that smoke exposure results in decreased host defence of the lung. Cigarette smoke applied to mice for 6–8 weeks resulted in a delayed rate of bacterial clearance compared with sham-exposed animals.4 Smoke decreases the function of mucociliary clearance,5 promotes adhesion of bacteria to the airway epithelia6 and affects the function of pulmonary host defence cells.7
Cigarette smoke is the most important risk factor for the development of COPD. COPD is a chronic progressive lung disease with a large medical and economic burden that develops in approximately 15–20% of all smokers. The pathogenesis of COPD is not clear. Chronic smoke exposure results in an inflammatory process characterised by the presence of neutrophils, macrophages and CD8+ lymphocytes. Detailed morphological analysis of tissue samples from patients with COPD revealed the presence of lymphoid aggregates containing follicles in the peripheral airways.8 Recurrent or chronic infections may be a driving force in the development of COPD. Smoking and COPD are related to bacterial colonisation of the pulmonary tract. Rising airway bacterial load is associated with greater airway inflammation and an accelerated decline in forced expiratory volume in 1 s.9 The diversity of the local microflora appears to change during the development of COPD.10 One hypothesis to describe the progression of COPD includes chronic or recurrent infections of the airways with subsequent activation of innate and adaptive immune mechanisms.
The innate host defence system of the lung is a first-line protection system. Antimicrobial peptides (AMPs) are important effector molecules of the innate immune system.11 The defensins and cathelicidins are the principal families of AMPs that are expressed in the respiratory tract. The β-defensins are mainly produced by epithelial cells and are secreted onto the airway surface where they have a broad spectrum of antimicrobial activity. In vitro studies and clinical studies show that the expression of several AMPs is induced by bacterial products and inflammatory mediators.11 12
The aim of the present study was to characterise the effect of smoking on functions of innate host defences and to correlate findings from clinical studies with results from in vitro experiments.
METHODS
Patients from CAPNETZ
CAPNETZ is the German Community Acquired Pneumonia Network that investigates the epidemiology, microbiology and clinical course of CAP (www.capnetz.de).13 Patients are identified by clinical signs and a positive lung radiograph. The programme was approved by local ethics committees and informed consent was obtained from the patients.
The material used for the study was obtained from 27 consecutive patients with CAP for whom the necessary samples (pharyngeal washing fluid, sputum) were available. The characteristics of the patients are summarised in table 1. Reformed smokers are defined as being abstinent for 12 months; other groups are never smokers and current smokers. Pharyngeal washing fluid was collected from 10 non-smoking and 10 smoking individuals with no current symptoms of infections by rinsing the mouth and gargling for 10 s. Pharyngeal washing fluid was used because the procedure can be performed non-invasively in sick patients and the material is suitable for the applied assays. It is commonly used to test for pathogens in lower airway infection.14
Measurement of human β-defensin-2 concentrations and Western blotting
Levels of the endogenous antibiotic peptide human β-defensin-2 (hBD-2) in pharyngeal washings obtained from patients were measured using an ELISA that detects hBD-2 in a range of 0.15–12.5 ng/ml. 96-well microtitre plates (Nunc, Wiesbaden, Germany) were coated with a polyclonal antibody15 against hBD-2 in 50 mM carbonate/bicarbonate buffer (pH 9.6) and the plates were blocked with 1% gelatin/PBS. 100 μl of samples were added into each well and incubated overnight at 4°C. A series of seven dilutions of recombinant hBD-215 was included as standard. Wells were washed and incubated with 100 μl of a second polyclonal antibody against hBD-2 (0.5 μg/ml AB 9871, Abcam, Cambridge, UK) and subsequently with 100 μl of a horseradish peroxidase (HRP)-labelled anti-goat antibody (1:40000, SigmaAldrich, Munich, Germany). Wells were washed again and 100 μl TMB + substrate solution (Dako, Carpinteria, California, USA) was added and incubated for 10 min. 100 μl stop solution (H2SO4, 3N) was added and the optical density of each well was determined using a microplate reader set to 450 nm. For test evaluation, spiking experiments were performed (recovery 79–81%).
Sputum from patients with pneumonia was analysed for hBD-2 by Western analysis. Samples were normalised to the weight of the initial sputum probe and measurements of protein before loading revealed equivalent protein quantities with a small variation (SD <15% of the mean). 10 μl complete protease inhibitor cocktail (Roche, Mannheim, Germany) was added per 1 mg sample. To homogenise the sample, 10 μl 100 mM 1,4-dithiothreit (DTT, Roth, Karlsruhe, Germany) were added per 100 mg of sputum. Five samples from smokers and five from non-smokers were pooled, extracted with 0.1% trifluoroacetic acid (TFA) and 10% acetonitrile. The material was applied to Sep-Pak Plus C18 cartridges (Waters, WAT020515, Milford, USA) and eluted with 0.1% TFA and 80% acetonitrile. The eluate was dried in a speed-vac concentrator and reconstituted in 20 μl distilled water. The extraction procedure of hBD-2 from sputum was validated by spiking experiments using commercially available hBD-2 peptide (Abcam). The detection limit was 2.5 ng hBD-2. The samples were separated on a 10–20% tris-tricine gel (Anamed, Darmstadt, Germany) and blotted on nitrocellulose membranes using Dunn-Carbonate buffer in a tank-blotting system (Bio-Rad Laboratories, Hercules, California, USA). For autoradiographic detection a polyclonal hBD-2 antibody15 and an HRP-labelled secondary rabbit-IgG antibody (GE-Healthcare, Little Chalfont, UK)) were used.
Cells and tissue culture, bacterial strain
Human bronchial epithelial cells were isolated from large airways resected during surgery and cultivated as submersed or air-liquid interface cultures as described previously.16 Donors (42, 39, 53 years of age) underwent lung transplantation for pulmonary fibrosis. The results did not differ between cells from the donors. The protocol was approved by the ethics committee of the University of Marburg and informed consent was obtained from the patients. Pseudomonas aeruginosa PAO1 bacteria were grown to an OD600 = 1.00 in LB media. For experiments with heat-inactivated bacteria, dilutions of 107 colony-forming units (CFU)/ml were prepared in phosphate-buffered saline (PBS) and incubated for 30 min at 96°C. Experiments with viable bacteria were performed using a dilution of 105 CFU/ml in PBS. P aeruginosa was used as a model organism because it is known to colonise the respiratory tract of patients with COPD and is sensitive to epithelial host defence factors. The number of CFUs used to infect cells was determined by plating an aliquot of the inoculum and varied between experiments.
Smoke exposure and infection models
Tissue cultures were exposed to volatile cigarette smoke as described previously.17 Briefly, tissue cultures were exposed to cigarette smoke for 15 min (3 cigarettes). After exposure the medium of the cultures was immediately replaced. Control cultures were incubated in the exposure chamber for the same time period without burning a cigarette. To determine the effects of cigarette smoke on the expression and release of hBD-2 and interleukin (IL)-8, heat-inactivated P aeruginosa PAO1 (107 CFU/ml in 30 μl) was applied to the apical side of the cultures for 8 h.
To investigate the effects of reactive oxygen species on the β-defensin expression in airway epithelium, cells were incubated with or without hydrogen peroxide for 1 h. After exposure the medium was replaced and the cells were stimulated with heat-inactivated P aeruginosa PAO1 (107 CFU/ml) for 8 h. Catalase (Sigma-Aldrich, Steinheim, Germany) was added to the culture medium at a concentration of 6400 U/ml immediately before and after smoke exposure.
Bacterial survival assays
The apical surface of differentiated epithelium was exposed to the smoke as described. After smoke exposure the cells were infected with 1×104–5 CFU viable P aeruginosa in 15 μl PBS. To determine the numbers of viable bacteria 6 h after infection, the apical surfaces of the cultures were washed with 100 μl PBS and serial dilutions were plated onto LB agar plates. 5 μg recombinant hBD-2 in 200 μl18 was applied to the apical surfaces of the cultures in some experiments based on concentrations calculated for airway secretions.19
Cytokine measurements
Levels of human IL-8 in tissue culture medium were determined by commercially available ELISAs according to the manufacturer’s instructions (R&D Systems, Wiesbaden-Nordenstadt, Germany).
Quantitative PCR
RNA isolation, reverse transcription and quantitative PCR were performed as described elsewhere.17 GAPDH primer (sense, 5′-GAAGGTGAAGGTCGGAGTC-3′; antisense, 5′-GAAGATGGTGATGGGATTTC-3′), hBD-1 primer (sense, 5′-GCCTCAGGTGGTAACTTTCTCA-3′; antisense 5′-GCGTCATTTCTTCTGGTCACT-3′) and hBD-2 primer (sense 5′-TCAGCTCCTGGTGAAGCTC-3′; antisense 5′-GGGCAAAAGACTGGATGACA-3′) were purchased from TIB Molbiol (Berlin, Germany).
Statistical methods
Values are displayed as mean (SD). Comparisons between groups were analysed by t test or ANOVA for experiments with more than two subgroups. Post hoc range tests were performed with the t test with Bonferroni adjustment. p Values <0.05 were considered statistically significant.
RESULTS
Epithelial defensin levels in smokers with CAP
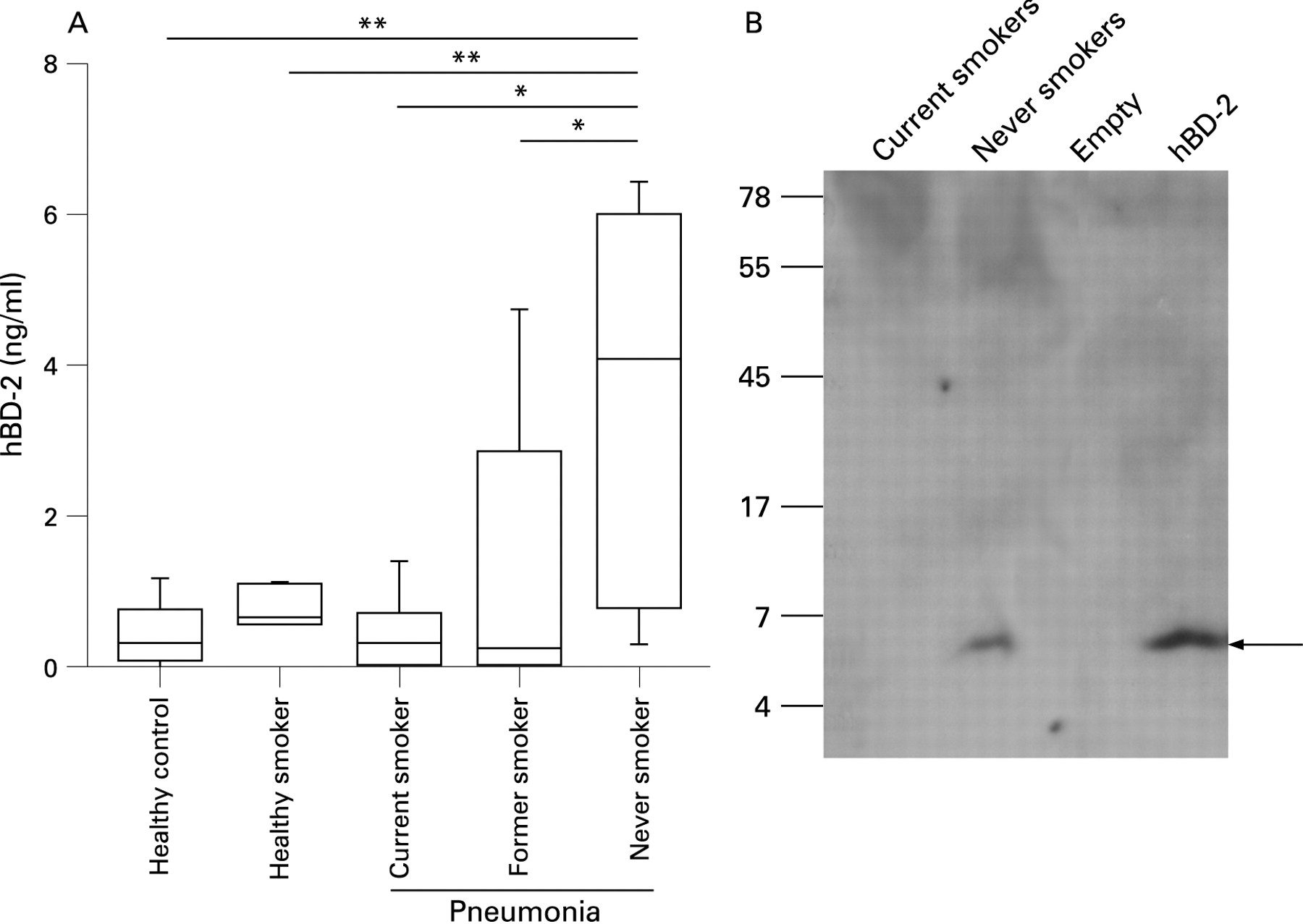
To determine whether smokers have decreased levels of mucosal AMPs in their airway secretions, we measured the amount of hBD-2 in pharyngeal flushing fluid and sputum from 27 patients with CAP by ELISA. Significantly decreased concentrations of hBD-2 were found in pharyngeal flushing fluid from current smokers and former smokers compared with never smokers (fig 1A). Healthy never-smoking and healthy current-smoking controls showed low levels of defensin. In a next step we analysed whether hBD-2 is present in the sputum of these patients. Owing to its physical properties, sputum could not be used for ELISA and therefore hBD-2 was analysed by Western blotting in extracts of pooled sputum samples. No sputum was available from healthy controls. hBD-2 was present in the sputum of never-smoking patients. In contrast, no peptide was found in the sputum of patients with CAP who were current smokers (fig 1B). It is likely that Western blotting is not sensitive enough to reveal small differences as determined by ELISA. These data show that epithelial antimicrobial defensins are induced during pneumonia and that current and former smoking suppresses this induction.
Effect of smoke exposure on the induction of epithelial defensin by bacteria
The effect of volatile cigarette smoke on the response of differentiated primary bronchial airway to P aeruginosa was investigated. Smoke-exposed cells showed decreased antimicrobial activity resulting in significantly increased numbers of viable bacteria after infection with P aeruginosa (fig 2A). Treatment of the cells with volatile cigarette smoke did not cause an increase in the release of lactate dehydrogenase (data not shown).

To investigate whether this defect of epithelial host defence after smoke exposure is correlated with decreased production of defensin, hBD-2 expression levels were measured after smoke exposure and in control cells. Smoke-exposed airway epithelium showed significantly suppressed hBD-2 mRNA induction in response to bacterial stimulation with heat-inactivated P aeruginosa (fig 2B). The expression of hBD-1 was not regulated under the applied conditions (fig 2B). The suppressed induction of hBD-2 expression correlated with a decrease in hBD-2 peptide secretion (fig 2C). Exogenously applied purified hBD-2 peptide significantly reduced the number of recovered bacteria from smoke-exposed and infected cells (fig 2D). Additional experiments showed that most viable bacteria were washed off by the procedures and that the bacteria attached to the cells did not influence the results (data not shown). Despite variations in the number of CFUs in the inoculum applied in individual experiments, the outcomes were qualitatively equivalent.
To test whether reactive oxygen species are involved in the mechanisms of defensin suppression, hydrogen peroxide was applied before exposing the airway epithelium to bacteria. Significant suppression of hBD-2 mRNA induction by heat-inactivated P aeruginosa occurred (fig 3A) without an increase in the release of lactate dehydrogenase from these cells (data not shown). The use of catalase as an antioxidant partly abrogated the effects of cigarette smoke on the induction of hBD-2 expression (fig 3B).

{kind=link}
{kind=link}
{kind=link}
To test whether smoke exposure resulted in a general decrease in cellular responses, we measured the release of IL-8 after exposure to smoke and heat-inactivated bacteria. The combination of smoke exposure and administration of P aeruginosa resulted in increased release of IL-8 (fig 3C).
DISCUSSION
The main finding of this study is that cigarette smoke suppresses the activation of the epithelial innate host defence system of the lung. Oxidative stress appears to play a role because hydrogen peroxide exposure mimics the suppressive effect of cigarette smoke and antioxidant treatment weakens this effect. Defensin appears to have a specific role in this mechanism because its expression correlates with host defence in tissue culture and clinical samples. External application of the peptide hBD-2 restored antimicrobial activity.
An increasing body of data supports the role of defensins in pulmonary host defence. In vitro experiments have shown wide antimicrobial activity20 and animal studies with mice deficient in mouse β-defensin 1 (mBD-1) found increased susceptibility to infection.21 22 The number of defensin gene copies varies between individuals (copy number variants). Individuals with low numbers of gene copies of the defensin locus are predisposed to Crohn’s disease.23 It is known that smoking alters the function of host defence components of the lung. Smoke-exposed animals also have increased inflammation but delayed clearance of P aeruginosa after infection.4 In addition, host defences against respiratory syncytial virus appear to be suppressed after exposure to smoke.24
The mechanism by which cigarette smoke causes a breach in the host defence is unclear. Cigarette smoke is known to inhibit the function of professional immune and host defence cells such as dendritic cells,25 T cells26 and alveolar macrophages.27 In this study we also show that the host defence function of airway epithelium is compromised by cigarette smoke. Smokers with pneumonia had significantly lower levels of hBD-2 in their mucosal secretion. Epithelial cells are the main cellular source of hBD-2 in the lung.15 Th2-biased inflammation during atopic dermatitis is correlated with decreased levels of AMPs and skin infections.28 We have shown that smoke exposure decreases epithelial host defences in the pulmonary system. In the study population, current smoking was associated with a lower age and less severe pneumonia. The mechanistic interaction between these factors and their impact on AMP expression is largely unclear.
Interestingly, the release of the proinflammatory cytokine IL-8 from epithelial cells is synergistically increased by the combination of smoke exposure and bacterial infection. In parallel, the host defence function and the expression of hBD-2 are decreased. Smokers have decreased levels of host defence peptide and the amount of inflammation in COPD is correlated with the magnitude of the bacterial load.9 Smoke is generally thought to affect proinflammatory pathways including MAP kinases p38, ERK1/2 and JNK or NF-κB.17 Acrolein is a major product of organic combustion and inhibits NF-κB activation by interaction with IkappaB kinase (IKK).29 The smoke component acrolein inhibits the baseline expression of IL-8 and hBD-2 in sinonasal epithelial cells.30 Smoke also inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppression of the activation of activator protein-1 in bronchial epithelial cells.31 Downregulation in TLR4 mRNA and protein expression in an epithelial cell line by cigarette smoke extract also could contribute to the effect of smoke on hBD-2 expression.32 Our data provide evidence that reactive oxygen species are involved in this process. Hydrogen peroxide could mimic the effect of smoke in epithelial host defence and catalase significantly increased the expression of hBD-2. The absence of lactate dehydrogenase release in our studies excluded frank cell necrosis. Apoptosis is a further mechanism that could result from smoke exposure.33 The increased production of IL-8 after smoke exposure showed that the effect of smoke on defensin expression is not due to an overall decrease in cell viability. The effect of smoke components on various signaling processes results in the differential expression of individuals genes as shown here for IL-8 and hBD-2.
The clinical implication of smoke-induced inhibition of pulmonary defences and the subsequent susceptibility to infection is obvious. In addition, a breach in innate host defence probably contributes to the pathogenesis of COPD. This chronic inflammatory lung disease is correlated with colonisation and increased bacterial load. A further feature of COPD is a self-perpetuating inflammatory process. Consistent with these findings, depression of hBD-2 induction was also found in former smokers who had stopped smoking more than 12 months previously. Smoke-induced decreased endogenous host defences might lead to an altered microflora of the lungs with increased numbers of microorganisms. Smoke is also known to break down the epithelial barrier that is essential for providing a physical barrier for microorganisms and limiting a host defence reaction.34 The combination of a broken epithelial barrier and the presence of microorganisms could results in a vicious cycle of inflammation, structural damage and infection. The contribution of antibacterial host defence molecules in this process has been suggested by an association study that showed that a genetic variation of human beta-defensin 1 (hBD-1) is found more frequently in patients with COPD than in controls.35
In conclusion, exposure to smoke reduces epithelial host defence, potentially resulting in increased susceptibility to colonisation and infection. This mechanism is probably involved in increased susceptibility to pulmonary infection.
Acknowledgments
The authors thank the CAPNETZ Study Group for making samples and data available for the study, and Thomas Damm for excellent technical support.
REFERENCES
Footnotes
Funding: Supported by grants from the Deutsche Forschungsgemeinschaft (Ba 1641/8-1;SBF/TR 22) to RB and from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) via CAPNETZ to RB (01KI0432), NS (01KI0426) and TW (01KI0429).
Competing interests: None.
Ethics approval: The study was approved by local ethics committees and informed consent was obtained from the patients.