Article Text

Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a fatal interstitial lung disease characterised by accumulation of activated (myo)fibroblasts and excessive extracellular matrix deposition. The enhanced accumulation of (myo)fibroblasts may be attributed, in part, to the process of transforming growth factor β1 (TGFβ1)-induced epithelial–mesenchymal transition (EMT), the phenotypic switching of epithelial to fibroblast-like cells. Although alveolar epithelial type II (ATII) cells have been shown to undergo EMT, the precise mediators and mechanisms remain to be resolved. The objective of this study is to investigate the role of SNAI transcription factors in the process of EMT and in IPF.
Methods: Using quantitative reverse transcription-PCR (RT-PCR), immunofluorescence, immunohistochemistry, western blotting, as well as gain- and loss-of-function studies and functional assays, the role of SNAI1 and SNAI2 in TGFβ1-induced EMT in ATII cells in vitro was assessed; and the expression of SNAI transcription factors was analysed in experimental and human IPF in vivo.
Results: TGFβ1 treatment increased the expression and nuclear accumulation of SNAI1 and SNAI2, in concert with induction of EMT in ATII cells. SNAI overexpression was sufficient to induce EMT, and small interfering RNA (siRNA)-mediated SNAI depletion attenuated TGFβ1-induced ATII cell migration and EMT. SNAI expression was elevated in experimental and human IPF and localised to hyperplastic ATII cells in vivo.
Conclusions: The results demonstrate that TGFβ1-induced EMT in ATII cells is essentially controlled by the expression and nuclear translocation of SNAI transcription factors. Increased SNAI1 and SNAI2 expression in experimental and human IPF in vivo suggests that SNAI-mediated EMT may contribute to the fibroblast pool in idiopathic pulmonary fibrosis.
Statistics from Altmetric.com
Idiopathic pulmonary fibrosis (IPF) is a progressive and lethal disorder of major concern due to its unresolved pathogenesis and limited responsiveness to currently available therapies.1 The hallmark lesions of IPF are fibroblast foci, which are sites featuring α-smooth muscle actin (αSMA)-positive, activated (myo)fibroblasts.2 3 Currently, three major theories attempt to explain the accumulation of activated (myo)fibroblasts in the lungs of patients with IPF. First, local fibroproliferation of resident pulmonary fibroblasts in response to fibrogenic cytokines and growth factors increases the fibroblast pool.4 Secondly, bone marrow-derived circulating fibrocytes trafficking to the lung may serve as progenitors for interstitial fibroblasts.5 6 7 8 9 Thirdly, alveolar epithelial type II (ATII) cells, via the process of epithelial–mesenchymal transition (EMT), can undergo a phenotypic, reversible switching to fibroblast-like cells.10 11 12
The orchestrated series of events during EMT includes remodelling of epithelial cell–cell and cell–matrix adhesion contacts, reorganisation of the actin cytoskeleton, induction of mesenchymal gene expression and the acquisition of motile capacity.13 During development and disease pathogenesis, EMT is under tight transcriptional control maintained by factors such as Twist, NF-κB, Rho, Rac, Snai or GSK-3β.13 14 The transcription factors eliciting EMT in IPF are yet to be identified. In this context, the zinc finger transcription factor SNAI1 (also called Snail) and SNAI2 (also called Slug) have been reported to act as regulators of EMT during development and disease, including cancer and organ fibrosis.15 16
Transforming growth factor β1 (TGFβ1) represents a main inducer and regulator of EMT in multiple organ systems, including the lung.10 17 While the ability of TGFβ1 to induce EMT has recently been described in ATII cells in vitro11 18 19 20 21 22 and in vivo in transgenic mice,12 the molecular mechanisms that control these dynamics and their precise role in other experimental models of lung fibrosis and IPF remain to be elucidated.
This study was performed to examine the hypothesis that TGFβ1 induces EMT via SNAI transcription factors in ATII cells and that SNAI transcription factors contribute to the development of IPF. In detail, we examined the characteristics of TGFβ1-induced EMT, modulated via SNAI transcription factor in vitro, and evaluated the expression pattern of SNAI transcription factors in experimental as well as human IPF in vivo.
Materials and methods
Reagents
The following antibodies were used in this study: anti-prosurfactant protein (proSP-C) (Chemicon International, Temecula, California, USA), anti-αSMA (Sigma, St Louis, Missouri, USA), anti-e-cadherin (ECAD) (BD Biosciences, San Jose, California, USA), anti-tight junction protein (TJP)1, anti-occludin (OCCL) (Zymed Laboratories, San Francisco, Calkifornia, USA), anti-SNAI1 and anti-α-tubulin (Santa Cruz Biotechnology, Santa Cruz, California, USA), anti-SNAI2 (Cell Signaling Technology, Beverly, Massachusetts, USA) and anti-SNAI1 (a gift from Dr Becker, Institute of Pathology, Technical University of Munich, Germany). Recombinant human TGFβ1 was purchased from R&D Systems (Minneapolis, Minnesota, USA).
Cell culture
The human lung epithelial cell line A549 (ATCC CCL-185; Manassas, Virginia, USA) was maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Pasching, Austria), supplemented with 10% fetal bovine serum (FBS; PAA Laboratories, Carlsbad, California, USA). Primary mouse ATII cells were isolated from adult male C57BL/6N mice as previously described.23
Human tissues
Lung tissue biopsies were obtained from 12 patients with IPF (mean age (SD) 51.3 (11.4) years; six females, six males) and nine transplant donors (serving as control subjects) (mean age 47.5 (13.9) years; four females, five males).24 The study protocol was approved by the Ethics Committee of the Justus-Liebig-University School of Medicine (AZ 31/93). Informed consent was obtained from each subject for the study protocol.
Animal tissues
All animal studies were performed in accordance with the guidelines of the Ethic’s Committee of University of Giessen School of Medicine and approved by the local authorities. Adult male C57BL/6N mice were treated with bleomycin and were sacrificed at the indicated time points.
RNA extraction and reverse transcription-PCR (RT-PCR)
Total RNA was extracted using RNeasy columns (Qiagen, Hilden, Germany) according to the manufacturer’s protocol, and PCR amplification was performed with human and mouse primers (Tables S1 and S2, online supplement).
Quantitative RT-PCR (qRT-PCR)
qRT-PCR was performed as previously described.23 All results were normalised to the relative expression of the constitutively expressed gene porphobilinogen deaminase. The relative transcript abundance of the target gene is expressed in ΔCt values (ΔCt = Ct reference–Ct target). Relative changes in transcript levels compared with controls are expressed as ΔΔCt values (ΔΔCt = ΔCt treated–ΔCt control). All ΔΔCt values correspond approximately to the binary logarithm of the fold change. The human and mouse primer sequences are depicted in Table S3 and S4, online supplement, respectively.
Small interfering RNA (siRNA) transfection
The siRNA oligonucleotides specific for human SNAI1 and SNAI2 mRNA (table 1) were obtained from Dharmacon Inc. (Lafayette, Indiana, USA). A549 cells were transiently transfected with 100 nM SNAI or non-specific siRNA using Lipofectamine2000 reagent (Invitrogen). At 4 h post-transfection, cells were treated with TGFβ1. Cells were lysed after 24 h and the efficiency of gene knock-down was monitored.
Small interfering RNA (siRNA) sequences
Migration assay
Cell migration was determined using Boyden chamber assay (ThinCerts Tissue Culture Inserts, 24-well, pore size 3.0 μm from Kremsmunster, Austria), as described previously.25 A549 cells were transfected with SNAI1, SNAI2 or non-specific siRNA at a total concentration of 75 nM, detached, and 5×104 cells were seeded into the Boyden chamber insert. Cells were cultured for 8 h to allow their attachment to the membrane and migration was induced by adding TGFβ1 (2 ng/ml) to the medium in the lower wells. After 24 h, cells were fixed and stained using crystal violet solution, and non-migrated cells were removed by cotton swabbing. The membranes were carefully separated from the insert wall, and optical densities of migrated cells were measured with a GS-800 Calibrated Densitometer and analysed with Quantity One software.
Statistical analysis of data
Values are presented as the mean (SEM). All ΔCt values obtained from qRT-PCR were analysed for normal distribution using the Shapiro–Wilk test, with the assignment of a normal distribution with p>0.05. All ΔΔCt values were analysed using the two-tailed, one-sample t test. Normality of data was confirmed using quantile–quantile plots. Intergroup differences of ΔCt values from patients and bleomycin-treated mice were derived using a one-tailed, two-sample t test. A one-way analysis of variance (ANOVA) with Tukey HSD post hoc test was for studies with more that two groups. A level of p<0.05 was considered statistically significant.
Further experimental procedures
The detailed procedures for western blotting, densitometric analysis, immunohistochemistry, immunofluorescence, laser-assisted microdissection and transfection of human SNAI1 and SNAI2, as well as the sequences of all primers used in this study, are provided in the online supplement.
Results
TGFβ1-induced EMT in ATII cells
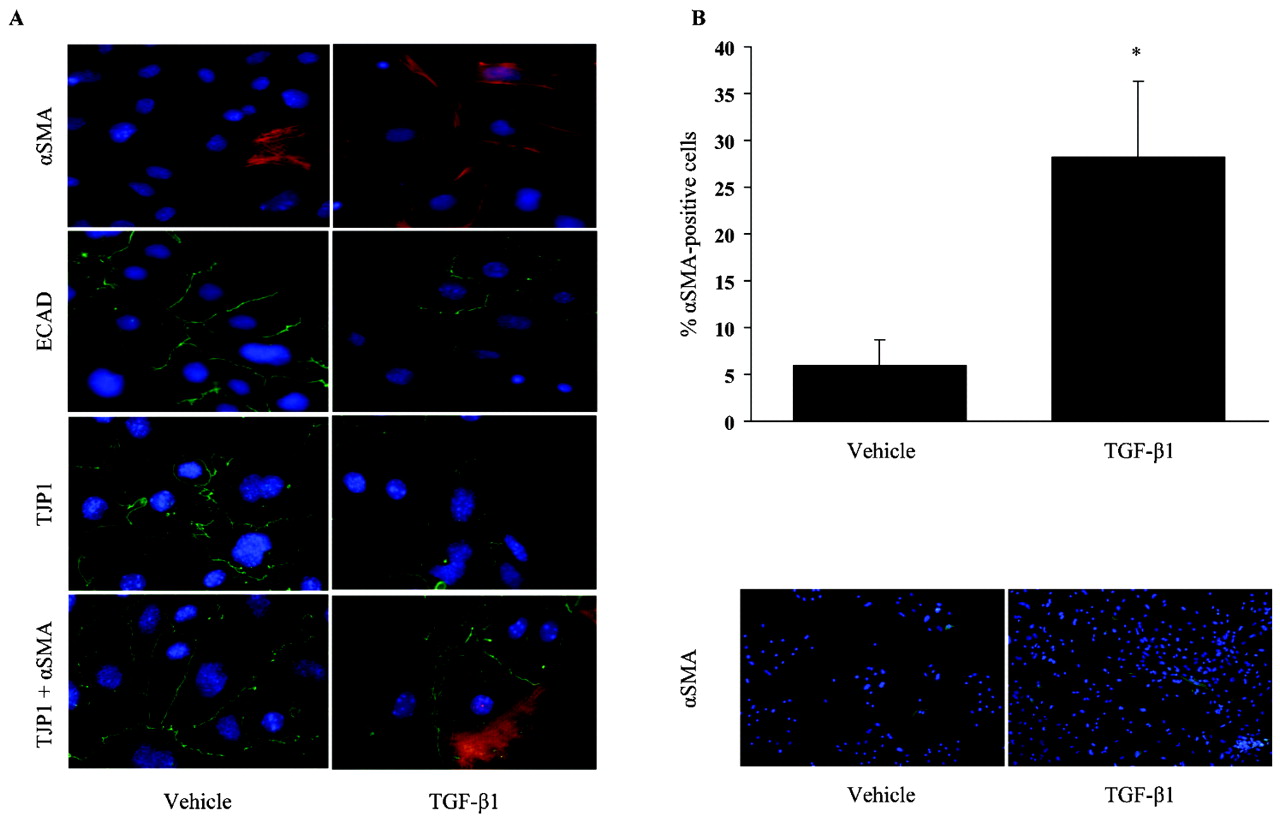
Initially, a comprehensive analysis of TGFβ1-induced EMT in ATII cells was performed. We assessed the gene and protein expression as well as the localisation of epithelial and mesenchymal markers in primary mouse ATII cells (Supplementary fig E1) and the human ATII cell line A549. TGFβ1 treatment for 24 h induced changes in αSMA, ECAD and TJP1 expression and/or localisation, indicative of EMT. While ECAD and TJP1 staining decreased, an appreciable number of cells expressing αSMA increased from 5.9% (2.8%) in vehicle-treated, to 28.2% (8.1%) in TGFβ1-treated cells (fig 1A,B). The presence of αSMA and TJP1 double-positive cells in TGF-β1-treated, but not in vehicle-treated cells, further corroborated the occurrence of TGFβ1-induced EMT in primary ATII cells (fig 1A, bottom panels).

Transforming growth factor β1 (TGFβ1)-induced epithelial–mesenchymal transition (EMT) in alveolar epithelial type II (ATII) cells. (A) Immunofluorescence detection of α-smooth muscle actin (αSMA), e-cadherin (ECAD) and tight junction protein 1 (TJP1) was performed after treatment with TGFβ1 (2 ng/ml) or vehicle for 24 h. Co-localisation of αSMA (red) and TJP1 (green) was assessed by immunofluorescence in ATII cells treated with TGFβ1 for 24 h. Nuclei are visible by 4′6-diamidino-2-phenylindole (DAPI) staining. Original magnification is ×40. (B) The quantification of the percentage of αSMA-positive cells 24 h after TGFβ1 (2 ng/ml) treatment. The original magnification of a representative figure showing immunofluorescence detection of αSMA (green) with TGFβ1 (2 ng/ml) or vehicle for 24 h is ×10. Results are representative of 10 independent experiments and data are expressed as mean (SEM); *p<0.05, n = 10.
SNAI transcriptions factor expression in TGFβ1-induced EMT in ATII cells
We subsequently quantified the mRNA expression patterns of several EMT marker genes in ATII cells, using qRT-PCR. ATII cells were treated with TGFβ1 in a dose- and time-dependent manner as indicated (fig 2, Supplementary fig E2). Although the magnitude of EMT marker gene expression in both cell types investigated differs, most markers were differentially regulated after 8 h. TGFβ1 treatment led to a decreased expression of the epithelial cell marker ECAD and occludin (OCCL), concomitant with increased expression of the mesenchymal marker vimentin (VIM) and αSMA, 8 h after treatment of ATII cells. Notably, VIM was strongly induced in primary ATII cells, but not in A549 cells (fig 2A,B). Importantly, we observed a significant upregulation of both SNAI1 and SNAI2, along with the alterations in EMT marker gene expression (fig 2A,B).

SNAI transcription factor expression in transforming growth factor β1 (TGFβ1)-induced epithelial–mesenchymal transition (EMT) in alveolar epithelial type II (ATII) cells. Using quantitative reverse transcription-PCR analysis, the expression pattern of the indicated EMT markers was detected in A549 (A) and primary mouse ATII cells (B), after TGFβ1 (2 ng/ml) treatment for 2 and 8 h, as indicated. Data are expressed as the mean (SEM); *p<0.05, n = 5. (C and D) Immunofluorescence analysis was performed using a primary antibody directed against SNAI1 and SNAI2 in both cells. The original magnification of the representative image from three independent experiments is ×40. ECAD, e-cadherin; OCCL, occludin; αSMA, α-smooth muscle actin; VIM, vimentin.
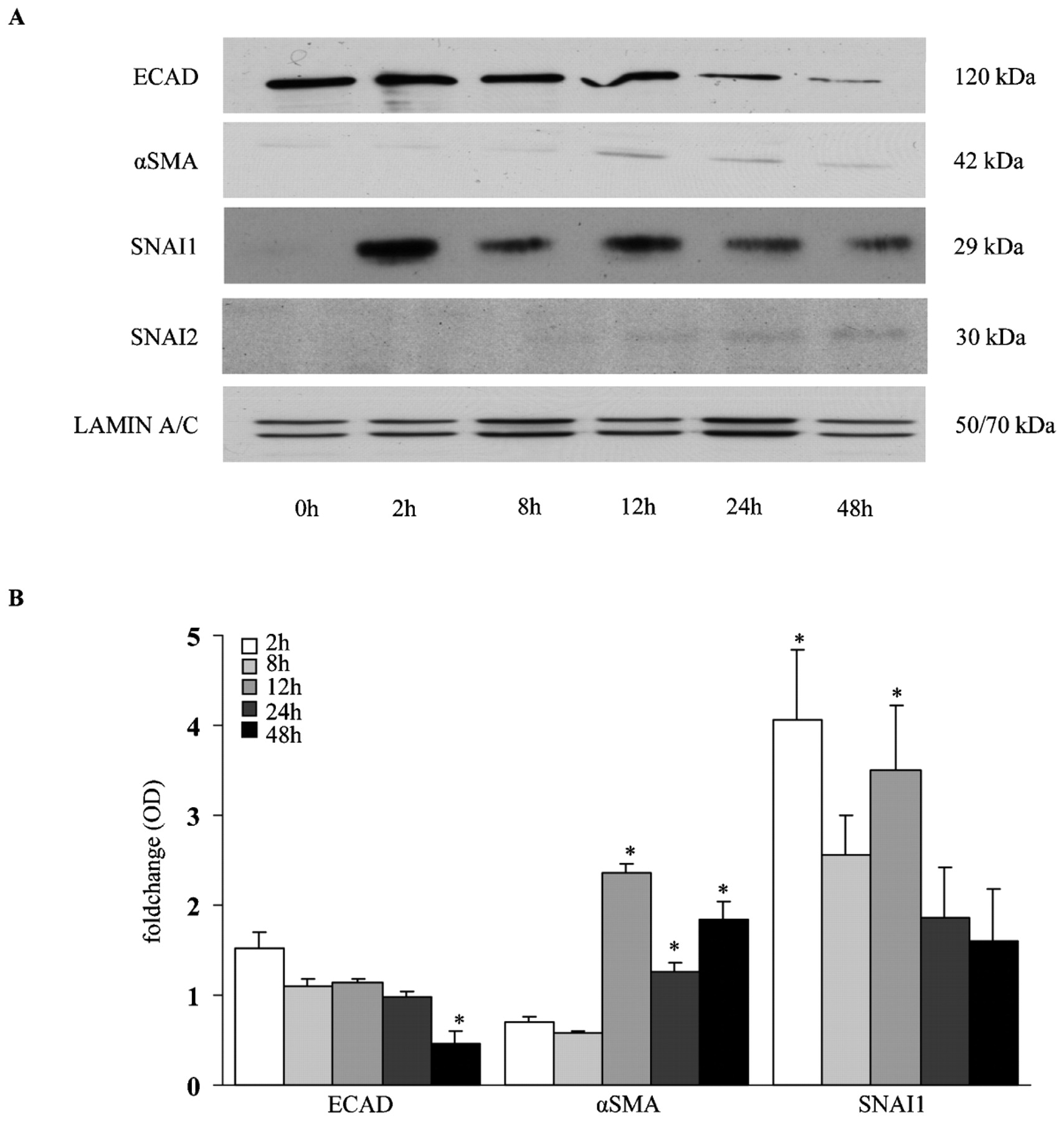
Since the SNAI transcription factors were regulated at the mRNA level, we next wanted to assess the protein expression and function in TGFβ1-induced EMT in ATII cells. Immunofluorescence analysis of TGFβ1-treated ATII cells revealed increased nuclear translocation of endogenous SNAI1 and SNAI2 upon TGFβ1 treatment in A549 and primary ATII cells, respectively (fig 2C,D, Supplementary fig E3). In addition, SNAI1 and SNAI2 protein expression was increased by TGFβ1 in a time-dependent manner, as assessed by western blot analysis. These changes were in concert with the time-dependent decrease of ECAD and increased αSMA expression in response to TGFβ1 treatment (fig 3A,B).

Protein expression of epithelial–mesenchymal transition (EMT) markers in transforming growth factor β1 (TGFβ1)-treated alveolar epithelial type II (ATII) cells. Protein expression of e-cadherin (ECAD), α-smooth muscle actin (αSMA), SNAI1 and SNAI2 was assessed by western blot analysis. A549 cells were treated with TGFβ1 (2 ng/ml) for several different times as indicated. Western blot analysis (A) was performed using primary antibody directed against ECAD, αSMA, SNAI1 and SNAI2. Lamin A/C served as a loading control. (B) Western blots were scanned and intensity is represented as fold change. Data are representative of three independent experiments and are expressed as the mean (SEM); *p<0.05, n = 3.
EMT induction by SNAI transcription factor overexpression in ATII cells
To investigate further whether SNAI has a functional role in inducing EMT in the lung, the full-length SNAI1 and SNAI2 cDNA were cloned into mammalian expression vectors and transiently transfected into A549 cells (transfection efficiency is depicted in Suppplementray fig E4). We examined the gene and protein expression pattern of different EMT marker post-SNAI transfection. SNAI1 overexpression led to a significantly increased αSMA expression (fig 4A), which was further confirmed on the protein level (fig 4B, Supplementary fig E5A). Interestingly, SNAI2 overexpression revealed significant changes on the gene and protein level of the epithelial cell targets ECAD and OCCL; however, no significant changes were observed for αSMA expression (fig 4C,D and Supplementary fig E5B). Taken together, overexpression of SNAI transcription factors is able to induce EMT in ATII cells, even in the absence of TGFβ1.
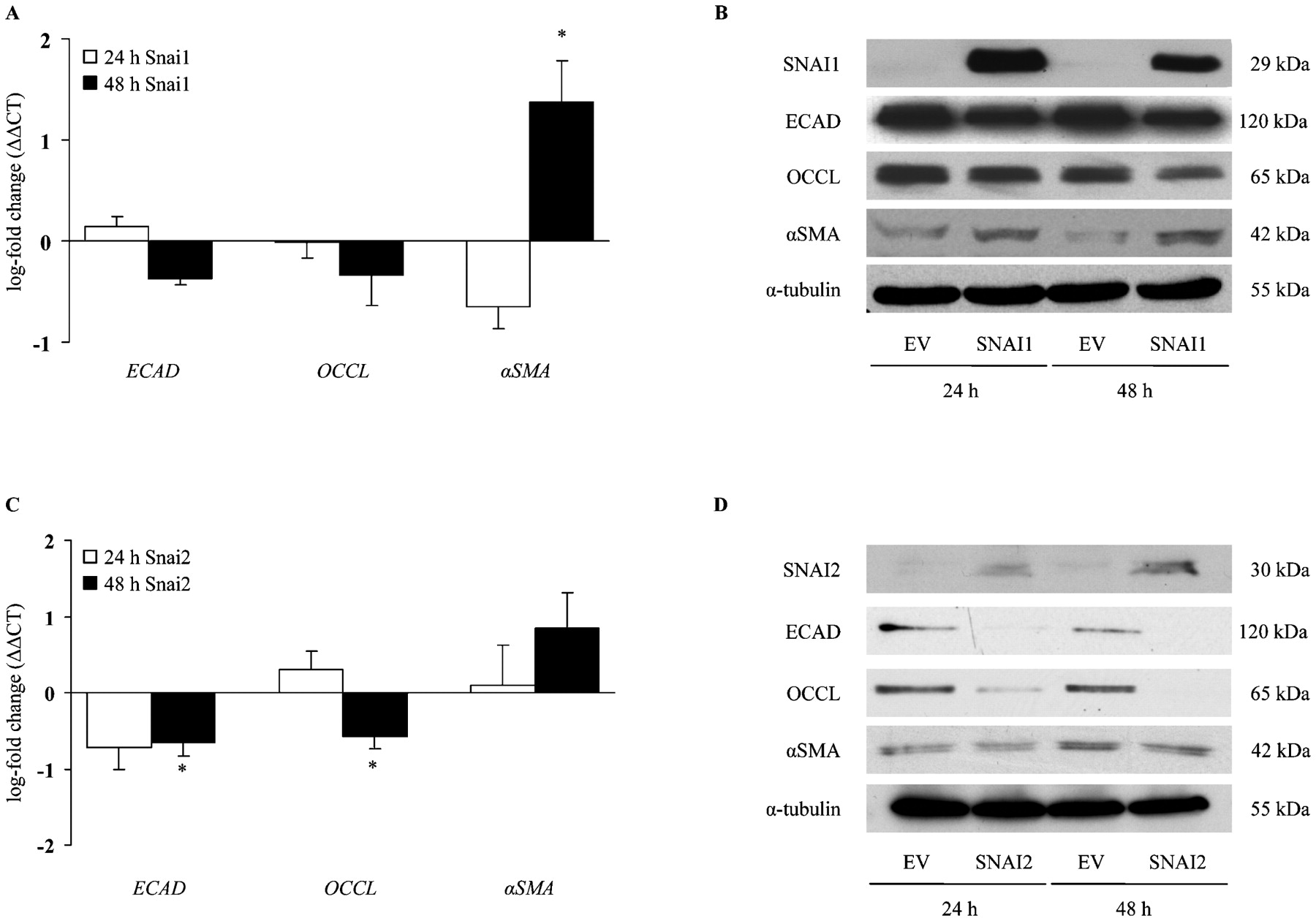
Epithelial–mesenchymal transition (EMT) induction by SNAI transcription factor overexpression in alveolar epithelial type II (ATII) cells. EMT marker expression was analysed by quantitative reverse transcription-PCR (A, C) and western blot (B, D). A549 cells overexpressing human SNAI1 (A, B) or SNAI2 (C, D) protein for 24 and 48 h; analysis was carried out by comparison with the empty vector (EV) control. The α-tubulin served as the loading control. Data are representative of five independent experiments and are expressed as the mean (SEM); * p<0.05, n = 5. ECAD, e-cadherin; OCCL, occludin; αSMA, α-smooth muscle actin.
Effect of SNAI transcription factor silencing on TGFβ1-induced EMT in ATII cells
In order to determine the effect of SNAI1 or SNAI2 on the process of TGFβ1-induced EMT in ATII cells, we next characterised SNAI mRNA knock-down using four different sequences of siRNA oligonucleotides targeting SNAI1 and SNAI2, respectively (Supplementary fig E6). Quantitative and semi-quantitative RT-PCR analysis revealed that three out of four SNAI1 (Supplementary fig E6A, B) and SNAI2 (Supplementary fig E6C, D) siRNA oligonucleotides were effective in reducing the respective mRNA level in response to TGFβ1 treatment. The most effective siRNA oligonucleotides targeting SNAI1 and SNAI2 were then used to examine the effect of SNAI depletion on TGFβ1-induced EMT. Both SNAI1 and SNAI2 depletion effectively attenuated TGF-β1-induced EMT in ATII cells (fig 5). SNAI1-depleted ATII cells exhibited a significantly attenuated decrease in OCCL and TJP1, as well as a reduced increase in αSMA mRNA levels, as assessed by qRT-PCR and semi-quantitative RT-PCR 24 h after TGFβ1 treatment compared with non-specific scrambled siRNA-treated ATII cells (fig 5A,B). Similarly, SNAI2 depletion in ATII cells demonstrated an increase in OCCL and TJP1 mRNA level compared with TGFβ1-treated non-specific scrambled siRNA-depleted ATII cells, whereas no significant changes have been observed in mesenchymal marker expression (fig 5C,D).

Effect of SNAI transcription factor silencing on transforming growth factor β1 (TGFβ1)-induced epithelial–mesenchymal transition in alveolar epithelial type II (ATII) cells. (A, C) Using quantitative reverse transcription-PCR (RT-PCR) analysis, the expression patterns of EMT marker genes was assessed in A549 cells after treatment with small interfering RNA (siRNA) against SNAI1 (A) and SNAI2 (C), with TGFβ1 exposures for 24 h and compared with control non-specific scrambled siRNA treatment with TGFβ1 exposures for 24 h. (B, D) Semi-quantitative RT-PCR analysis; the expression patterns of EMT marker genes was detected in A549 cells after siRNA treatment against SNAI1 (B) and SNAI2 (D), with TGFβ1 exposures for 24 h (lane 4) and compared with controls (1) non-specific scrambled siRNA treatment with (lane 3) or without (lane 1) TGFβ1 for 24 h and (2) siRNA against SNAI without TGFβ1 treatment (lane2). HSPA8 (heat shock 70 kDa protein 8) was employed as loading control. Data are representative of three independent experiments and are expressed as mean (SEM); * p<0.05, n = 3. ECAD, e-cadherin; OCCL, occludin; scr; scrambled siRNA oligonucleotide; αSMA, α-smooth muscle actin; TJP1, tight junction protein 1; VIM, vimentin.
Effect of SNAI transcription factor silencing on TGFβ1-induced migration in ATII cells
One of the key requirements for the complete process of EMT is the capability of ATII cells to migrate. TGFβ1 induces ATII cell migration in a dose-dependent manner (Supplementary fig E7). In order to elucidate the impact of SNAI transcription factor on TGFβ1-induced ATII cell migration, we performed an in vitro migration assay on SNAI1- and SNAI2-depleted cells stimulated with TGFβ1. After 24 h of TGFβ1 treatment, cells transfected with non-specific siRNA exhibited a fourfold induction of cell migration (fig 6). Both SNAI1- and SNAI2-depleted cells exhibited significant reduced migration under baseline and TGFβ1-induced conditions (fig 6).

Effect of SNAI transcription factor silencing on transforming growth factor β1 (TGFβ1)-induced migration in alveolar epithelial type II (ATII) cells. The small interfering RNA (siRNA)-treated A549 cells were treated or not with TGFβ1 for 24 h. The relative migration potential was assessed using Boyden chamber assay. scr; scrambled siRNA oligonucleotide. Membranes were scanned and the intensity is represented as bars. Data are representative of three independent experiments and are expressed as mean (SEM); * p<0.05, n = 3.
SNAI transcription factor expression in experimental lung fibrosis
To analyse whether expression of SNAI transcription factors was regulated during experimental lung fibrosis, we subjected mice to bleomycin treatment for 7 or 14 days. On the mRNA level, a significant increase in SNAI1 has been observed in lung homogenates from bleomycin-treated mice compared with saline-treated mice, as assessed by qRT-PCR analysis (fig 7A). Interestingly, significantly elevated levels of both SNAI1 and SNAI2 were observed in freshly isolated ATII cells from bleomycin-treated lungs compared with ATII cells from saline-treated lungs, as early as 7 days after treatment (fig 7B). Using immunohistochemistry, we next determined the localisation of SNAI1 and SNAI2 in bleomycin-treated lungs. While SNAI1 was predominantly localised to perivascular mesenchymal cells and alveolar macrophages in saline-treated lungs, it also demonstrated localisation in the epithelium and, particularly, in subepithelial areas, after bleomycin treatment (fig 7C). Although little or no staining for SNAI2 was detectable in saline-treated lungs, weak SNAI2 staining localised to the lung interstitium after bleomycin treatment (fig 7C). αSMA served as a well-characterised marker of the lung interstitium after bleomycin treatment.

SNAI transcription factor expression in experimental lung fibrosis. Mice were exposed to bleomycin, and lungs were harvested after 7 or 14 days, as indicated. (A) RNA was isolated and quantitative reverse transcription-PCR (qRT-PCR) was performed for SNAI genes in 7 or 14 day bleomycin- or saline-treated lung homogenates. (B) SNAI gene expression was quantified in primary alveolar epithelial type II (ATII) cells freshly isolated from 7 or 14 day bleomycin-treated lungs, using qRT-PCR. Data are representative of five independent experiments and are expressed as mean (SEM); * p<0.05, n = 5. (C) Immunohistochemical analysis of α-smooth muscle actin (αSMA), SNAI1 and SNAI2 localisation was performed in paraffin-embedded tissue from bleomycin- or saline-treated lungs after 14 days.
SNAI transcription factor expression in IPF
Finally, we asked whether SNAI expression was also increased in the lungs of patients with IPF. To this end, we determined the mRNA expression in lung homogenates obtained from patients with IPF and transplant donors. The qRT-PCR analysis revealed increased levels of SNAI2 gene expression in both lung homogenates and microdissected alveolar septae from patients with IPF compared with transplant donors (fig 8A,B). Immunohistochemistry further confirmed the expression and localisation of SNAI1 and SNAI2 predominantly in hyperplastic alveolar epithelial cells in IPF, whereas only weak SNAI1 and SNAI2 expression was detected in lung specimens of transplant donors (fig 8C). Western blot analysis further corroborated the elevated protein level of both SNAI1 and SNAI2 in lung homogenates from patients with IPF compared with transplant donors (fig 8D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SNAI transcription factor expression in idiopathic pulmoray fibrosis (IPF). (A) Gene expression analysis of SNAI1 and SNAI2 genes was performed by quantitative reverse transcription-PCR (qRT-PCR) analysis of total RNA derived from lung homogenates of 12 patients with IPF and nine transplant donors. (B) SNAI1 and SNAI2 mRNA levels were quantified using qRT-PCR in laser-assisted microdissected septae from five patients with IPF and five transplant donors. Data are expressed as the mean (SEM); *p<0.05, n = 5. (C) Immunohistochemical analysis of SNAI1 and SNAI2 localisation was performed in a paraffin-embedded lung specimen from patients with IPF or transplant donors. (D) Western blot analysis of SNAI1 and SNAI2 was performed three times in lung homogenates obtained from six patients with IPF and six transplant donors. Lamin A/C served as the loading control.
Discussion
IPF is the most common form of idiopathic interstitial pneumonias (IIPs), which exhibits a poor prognosis and unresponsiveness to currently available therapies, reflecting our limited understanding of the basic mechanisms and mediators implicated in the pathogenesis of this disease.1 26 27 While the initial injury in IPF is affecting the alveolar epithelium, the interstitial fibroblast/activated (myo)fibroblast represents the key effector cell accumulating in fibroblast foci and responsible for the increased extracellular matrix deposition that is characteristic for IPF.28 29 30 The EMT, a process where alveolar epithelial cells turn into fibroblast-like cells, has recently been reported to serve as a source for the fibroblast pool in tissue fibrosis of the liver, kidney and lung.11 12 31 EMT was initially described in embryonic development.14 The orchestrated series of events during EMT involves changes in cell polarity, loss of epithelial cell markers, induction of mesenchymal gene expression and enhanced cell migration.13 TGFβ1, known as a key profibrotic growth factor, has emerged as a potent inducer of EMT.17 32 In the present study, we report that SNAI transcription factors are key regulators of TGFβ1-induced EMT in the lung. Elevated expression of SNAI1 and SNAI2 was observed in ATII cells in response to TGFβ1 in vitro. Depletion of SNAI1 and SNAI2 using siRNA knock-down in ATII cells inhibited TGFβ1-induced alterations in EMT marker gene expression and ATII cell migration in response to TGFβ1. Interestingly, ectopic overexpression of SNAI transcription factors promotes EMT even in the absence of TGFβ1. Finally, an increased level of SNAI1 and SNAI2 in experimental and human IPF in vivo further indicated a significant contribution of SNAI transcriptions factors to the process of EMT in lung fibrosis.
At the onset of our studies, we performed a detailed analysis of the occurrence of EMT in ATII cells in response to TGFβ1, determining marker gene and protein expression and localisation in primary mouse ATII cells and A549 cells. Upon exposure to TGFβ1, ATII cells demonstrated an increased expression of mesenchymal markers (such as αSMA) with a corresponding decrease in epithelial markers (ECAD and TJP1), suggestive of EMT. EMT was further corroborated by the presence of αSMA and TJP1 double-positive cells in TGFβ1-treated, but not in untreated cells. These results are in accordance with previous studies, reporting that TGFβ1 induces EMT in lung epithelial cells in vitro.11 19 20 21 Recently, TGFβ1-induced EMT has also been demonstrated in vivo in a triple transgenic mouse model.12
Several regulatory molecules have been implicated in the process of EMT.13 17 Here, we demonstrated increased expression and nuclear translocation of the zinc finger transcription factors SNAI1 and SNAI2 along with EMT in ATII cells. Elevated levels of SNAI1 mRNA have been reported in TGFβ1-treated A549 cells,22 which has been further confirmed in this study. In addition, SNAI1 induction by TGFβ1 has been recently demonstrated in MDCK cells, a canine renal epithelial cell line.33 Furthermore, it has been demonstrated that SNAI1-deficient mice die at the gastrulation stage,34 because of their inability to undergo EMT, reinforcing the importance of the SNAI transcription factors in the process of embryonic development.
The impact of SNAI-mediated EMT in pathophysiological conditions such as cancer or tissue fibrosis, however, is less well substantiated and requires further investigations. Recently, high SNAI expression has been associated with poor prognosis and tumour recurrence in patients with lung cancer.35 In addition, SNAI1 has been reported to induce chemoresistance of cancer cells.36 In tissue fibrosis, SNAI-mediated EMT has been proposed to be involved in kidney fibrosis.37 The comprehensive analysis presented herein strongly suggests that SNAI1 and SNAI2 are essential mediators involved in the initiation and perpetuation of TGFβ1-mediated EMT in experimental and human IPF, but by no means proof of EMT in IPF in vivo. Since we were able to inhibit EMT in ATII cells with siRNAs targeting SNAI1 and SNAI2, it will have to be demonstrated whether in vivo interference with these factors may lead to an attenuation of fibrosis. We also present evidence that both SNAI1 and SNAI2 induce EMT even in the absence of TGFβ1, hence recapitulating their role as potent inducers of EMT. It has to be pointed out that SNAI1 and SNAI2 may differ in their respective target genes. Our results suggest that SNAI1 preferably regulated αSMA, whereas SNAI2 alters epithelial targets. This is of special interest, as cell-specific SNAI expression in vivo may lead to distinct cell fates. Given that SNAI2 was the dominant transcription factor regulated in human IPF tissue, we propose that some ATII cells undergoing EMT in IPF may exhibit an αSMA-negative fibroblast phenotype mediated by SNAI2.
In IPF, recent studies were able to demonstrate evidence of EMT in lung tissue biopsies, suggesting that this process contributes to the increased pool of (myo)fibroblasts in lung fibrosis,11 12 as well as in allografts after lung transplantation.38 In contrast, another recent study failed to supply evidence for EMT in pulmonary fibrosis.39 This discrepancy may be due to the transient nature and complexity of the EMT process, and further highlights the challenge in this research field.
Taken together, we demonstrated that SNAI transcription factors mediate EMT in ATII cells in vitro and their expression is increased in experimental and human IPF in vivo. We speculate that EMT is an early event in IPF and that activation and nuclear translocation of SNAI transcription factors constitute an important early regulator of EMT in ATII cells.
Acknowledgments
We thank Dr Becker (Institute of Pathology, Technical University of Munich, Germany) for the generous gift of anti-rat monoclonal SNAI1 antibody. We are indebted to all members of the Eickelberg Lab for stimulating discussions, and Andreas Jahn and Simone Becker for excellent technical assistance.
REFERENCES
Footnotes
Funding The authors are supported by the Helmholtz Association, the German Research Foundation (DFG) KliFo 118, the International Graduate Program “Signaling Mechanisms of Lung Physiology and Disease” GRK1062, and a career development award by the University of Giessen School of Medicine to M.K.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Ethics approval The study protocol was approved by the Ethics Committee of the Justus-Liebig-University School of Medicine (AZ 31/93)
▸ Additional tables, figures and experimental procedures are published online only at http://thorax.bmj.com/content/vo64/issue12