Article Text

Statistics from Altmetric.com
S92 PULMONARY ENDOTHELIAL CELL SECRETION OF FACTOR VIII
1CL Shovlin, 2R Manning, 2G Angus, 1M Laffan, 1G Birdsey, 2FA Mauri. 1Imperial College London, London, UK, 2Hammersmith Hospital, Imperial College Healthcare NHS Trust, London, UK
Introduction: Activated factor (F)VIII is responsible for sustained intravascular generation of thrombin via its role as a cofactor for FIXa in the coagulation cascade. Elevated plasma levels of FVIII are a strong predictor of recurrent venous thromboses/pulmonary emboli and are associated with thromboembolic pulmonary hypertension. The sources of plasma FVIII have remained elusive. Having demonstrated that FVIII levels are elevated in hereditary haemorrhagic telangiactasia associated with pulmonary arteriovenous malformations (Shovlin et al. Thromb Haemost 2007), we tested the hypothesis that human pulmonary artery endothelial cells (HPAEC) secrete FVIII and compared expression patterns with three other primary endothelial cell types under basal and stimulated conditions.
Methods: Primary human endothelial cells (Promocell) were cultured in individual wells of BD Falcon Culture Slides, fixed and permeablised with methanol. Wells were treated with one of four hybridoma-derived murine anti-human FVIII:Ag monoclonal antibodies and rabbit-raised anti-von Willebrand Factor (vWF), anti-calnexin or anti-COPII. Primary antibodies were detected with Alexa-Fluor labelled anti-rabbit or anti-mouse IgG. Stained cells covered with Vectashield were imaged on an LSM 510 Zeiss inverted fluorescence confocal microscope using sequential acquisition and LSM Image Browser software. Prior to immunohistochemistry of lung tissue blocks, antigen retrieval methods were optimised using paraffin-embedded formalin-fixed blocks derived from cultured HPAEC. FVIII secretion into conditioned media was quantified by ELISA (Immunobind, Axis-Shield) using manufacturer’s protocols.
Results: All four anti-FVIII antibodies to both heavy and light FVIII chains demonstrated significant reactivity in HPAEC. In contrast to reported retention of endogenous FVIII in hepatocyte endoplasmic reticulum (ER) (Becker et al. Thromb Haemost 2004), in HPAEC there was limited colocalisation with calnexin. Colocalisation was observed with COPII demonstrating ER–Golgi traffic and in some cells, with vWF-containing Weibel Palade bodies. FVIII expression differed markedly between different endothelial cell types. Following 48 h of culture, secretion of FVIII into conditioned media was demonstrable. Immunohistochemistry confirmed FVIII staining of CD31-positive pulmonary endothelial cells in surgically resected lung tissue.
Conclusion: Pulmonary endothelial cells secretion of factor VIII may be relevant to intravascular prothrombotic states.
S93 INVESTIGATION OF THE PROTEASE/ANTIPROTEASE BALANCE IN THE BRONCHOALVEOLAR LAVAGE FLUID OF PATIENTS WITH WEGENER’S GRANULOMATOSIS
1DG Atherton, 2AG Richter, 1JA Warner, 2DR Thickett. 1Southampton University Medical School, Southampton, UK, 2University of Birmingham, Birmingham, UK
Wegener’s granulomatosis (WG) is a multisystem necrotising vasculitis of small arteries and veins that is characterised by granuloma formation. Recent studies have suggested that significant numbers of WG patients have abnormal lung function particularly of the small airways. This is thought to be due to the abnormal airway remodelling seen in this condition. Remodelling of the airways may be related to an imbalance in the protease/anti-protease ratio such as is thought to be present in chronic obstructive pulmonary disease (COPD). The aim of this study was to determine whether there is an imbalance in the MMP-9/TIMP-1 axis in these patients.

Method: Bronchoalveolar lavage fluid (BALF) samples from 31 WG patients were analysed and compared with six healthy controls. MMP-9 and TIMP-1 concentrations were determined by ELISA and the molecular profile of MMP-9 was analysed by zymography.
Results: MMP-9 was significantly higher in patients with WG than in controls, with median values of 6.31 vs 0.38 ng/ml (p<0.05). The ratio of MMP-9/TIMP-1 was also elevated in WG patients (median 0.65 vs 0.08, p<0.05), respectively. Interestingly, a large variation was seen throughout the WG samples that remained even once MMP-9 was corrected for total protein. Patients in all disease stages (acute, relapse and remission) had elevated MMP-9/TIMP-1 ratios when compared with controls with median values of 0.27, 0.88 and 0.69, respectively. MMP-9 levels positively correlated with neutrophil count in the BALF (Rho 0.493 and p<0.05). WG patients with bronchiectasis on HRCT had significantly higher levels of MMP-9 than those without (median 251 vs 4.37 ng/ml, p<0.05). Zymography showed that both active and pro-MMP-9 are elevated in WG patients (see fig).
Conclusions: The MMP-9/TIMP-1 balance in the WG lung appears to be dysregulated possibly due to excessive protease release from activated neutrophils that is not counterbalanced by antiprotease activity. This suggests a net matrix degrading effect that may be contributing to the lung pathology seen in this condition. As this dysregulation continues even once patients enter remission, this suggests that ongoing matrix remodelling could contribute to the long-term lung impairment seen in WG. This highlights the possibility for a new area of pharmacological treatment of this condition.
S94 CIRCULATING CYTOKINE PROFILES IN PATIENTS WITH CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION AND RESPONSE TO PULMONARY ENDARTERECTOMY
1E Soon, 2AM Holmes, 1M Toshner, 1C Treacy, 3J Suntharalingam, 1K Sheares, 2DC Budd, 1J Pepke-Zaba, 1NW Morrell. 1Papworth Hospital NHS Trust, Cambridge, UK, 2Respiratory Disease Area, Novartis Horsham Research Centre, Sussex, UK, 3Royal United Hospital NHS Trust, Bath, UK
Introduction: Mortality in chronic thromboembolic pulmonary hypertension (CTEPH) is greatly improved if patients are suitable for and treated with pulmonary thromboendarterectomy (PTE).1 It has been shown that levels of cytokines involved in the inflammatory process change immediately after PTE.2 This study aims to characterise the overall cytokine profile in patients pre and 3 months post-PTE.
Methods: 34 patients with CTEPH suitable for PTE consented for blood to be drawn prior to PTE and 3 months afterwards. ELISA were used to measure IL-1b, IL-2, IL-4, IL-5, IL-8, IL-10, IL-12p70 and IL-13; tumour necrosis factor alpha and IFN-γ. All patients were assessed pre and post-PTE with right heart catheterisation, imaging and 6-minute walks. Twenty-four healthy volunteers served as controls.
Results: These are summarised in the table. Patients with CTEPH had increased circulating levels of IL-1b, IL-2, IL-4 and IL-8 compared with controls. Levels of IL-2 and IL-8 fell significantly 3 months post-PTE.
Conclusions: The overall cytokine profile of these patients differs from healthy controls and changes significantly post-PTE. These patterns of cytokine changes may serve as useful biological markers of severity of pulmonary hypertension and response to surgery.
S95 EPOPROSTENOL USE IN A NATIONAL PULMONARY HYPERTENSION CENTRE FROM 1997 TO 2007
AC Church, D Sanabria, AJ Peacock, MK Johnson. Scottish Pulmonary Vascular Unit, Glasgow, UK
Introduction: Recently the cost-effectiveness of epoprostenol in pulmonary arterial hypertension (PAH) has been called into question (NICE appraisal document 2008). To assess the impact that withdrawal of intravenous epoprostenol in 1997 would have had, we retrospectively reviewed its use in a well defined population of patients with PAH.
Methods: Patients who were treated with intravenous epoprostenol from 1997 to 2007 were identified from the Scottish Pulmonary Vascular Unit database. Case notes were then reviewed to determine the following parameters: (1) reason for initiation of epoprostenol; (2) outcome after 12 months of treatment as assessed by changes in conventional outcome measures (when data were available) and survival greater than 12 months; (3) outcome in those patients whose treatment with epoprostenol commenced after licensing of oral disease targeted therapy in 2002.
Results: 50 (20%) of the patients from a total population of 252 were identified as having been on epoprostenol. The majority were idiopathic PAH (50%) although all aetiologies in group 1 of the Venice classification were represented. Epoprostenol was started first line for severe disease in 26% (n = 13), second line due to clinical deterioration in 46% (n = 23), as short-term rescue therapy in 6% (n = 3) and as the only available treatment pre-2002 in 22% (n = 11). Taking the subset of patients following the introduction of oral agents in 2002, 15% (30 out of 199) required epoprostenol: 57% improved, 30% died, 10% stabilised and 3% were intolerant. In total, 20 out of 30 patients either improved or stabilised over 12 months. Overall improvements in functional class, 6-minute walk distance (mean increase of 57.9 m (148.5 to −30 m)), NTproBNP (mean fall 1819 pg/ml) and quality of life (CAMPHOR) were observed. Included were 10 patients who deteriorated on oral agents and required epoprostenol: five improved, three died, one stabilised and one was intolerant.
Conclusions: In our centre the number of patients requiring epoprostenol represents 20% of the overall PAH population. Patients who deteriorate on oral agents can have their deterioration stabilised or reversed with the addition of epoprostenol. Epoprostenol should remain an important and available treatment for PAH. This should have implications for the forthcoming NICE review on the treatment of PAH.
S96 ISOLATION OF MULTIPLE FVIII TRANSCRIPTS IN PULMONARY AND SYSTEMIC VASCULAR ENDOTHELIAL CELLS
1GN Okoli, 1FS Govani, 2IG Mollett, 1CL Shovlin. 1Imperial College London, London, UK, 2Instituto de Medicina Molecular, Lisbon, Portugal
Introduction: Factor VIII (FVIII) is an essential component of the haemostatic network, mutated in patients with haemophilia A and implicated in the pathogenesis of chronic thromboembolic pulmonary hypertension (CEPH) and recurrent venous thromboembolism (VTE). The FVIII gene was known to encode two messenger RNA isoforms, the major isoform that encodes the protein that circulates in plasma and associates with von Willebrand factor and a smaller poorly characterised isoform. Following definition of FVIII protein expression, we hypothesised that further FVIII isoforms may be expressed in vascular endothelial cells.
Methods: RNA was extracted using Tri Reagent (Sigma) preparations of primary human endothelial cells (pulmonary artery, pulmonary microvascular, dermal microvascular and human umbilical vein endothelial cells). Complementary DNA were synthesised using SuperScript II. Following identification of FVIII isoforms in deposited mRNA and EST sequence databases by gene modelling algorithms, oligonucleotide PCR primers were designed to differentiate between putative isoforms. RT–PCR products were purified by gel excision or treated with an ExoSAP mix comprising 0.8 u of ExoI and 0.7 u of SAP and sequenced on an ABI 3730x1 DNA analyser and results analysed using FinchTV V1 4.0 software (Geospiza). Confirmation of FVIII transcript identity was obtained by Blastn (NCBI) and sequence alignments performed.
Results: Overall, five different transcripts were identified in the endothelial cell series. Two corresponded to the known major and minor isoforms. There were three other transcribed isoforms that are not present in Ensembl or NCBI databases. Isoform expression appeared to vary between endothelial cell types, and endothelial cell culture conditions. Semiquantitative RT–PCR suggested that the full length transcript was not the predominant isoform in most endothelial cells. Formal quantitative evaluation of vascular bed-specific expression is underway.
Conclusions: We have identified multiple factor VIII transcript isoforms in endothelial cells. Transcription-associated gene regulation and alternative pre-mRNA splicing of FVIII could have significant implications for the pathogenesis of thromboembolic diseases.
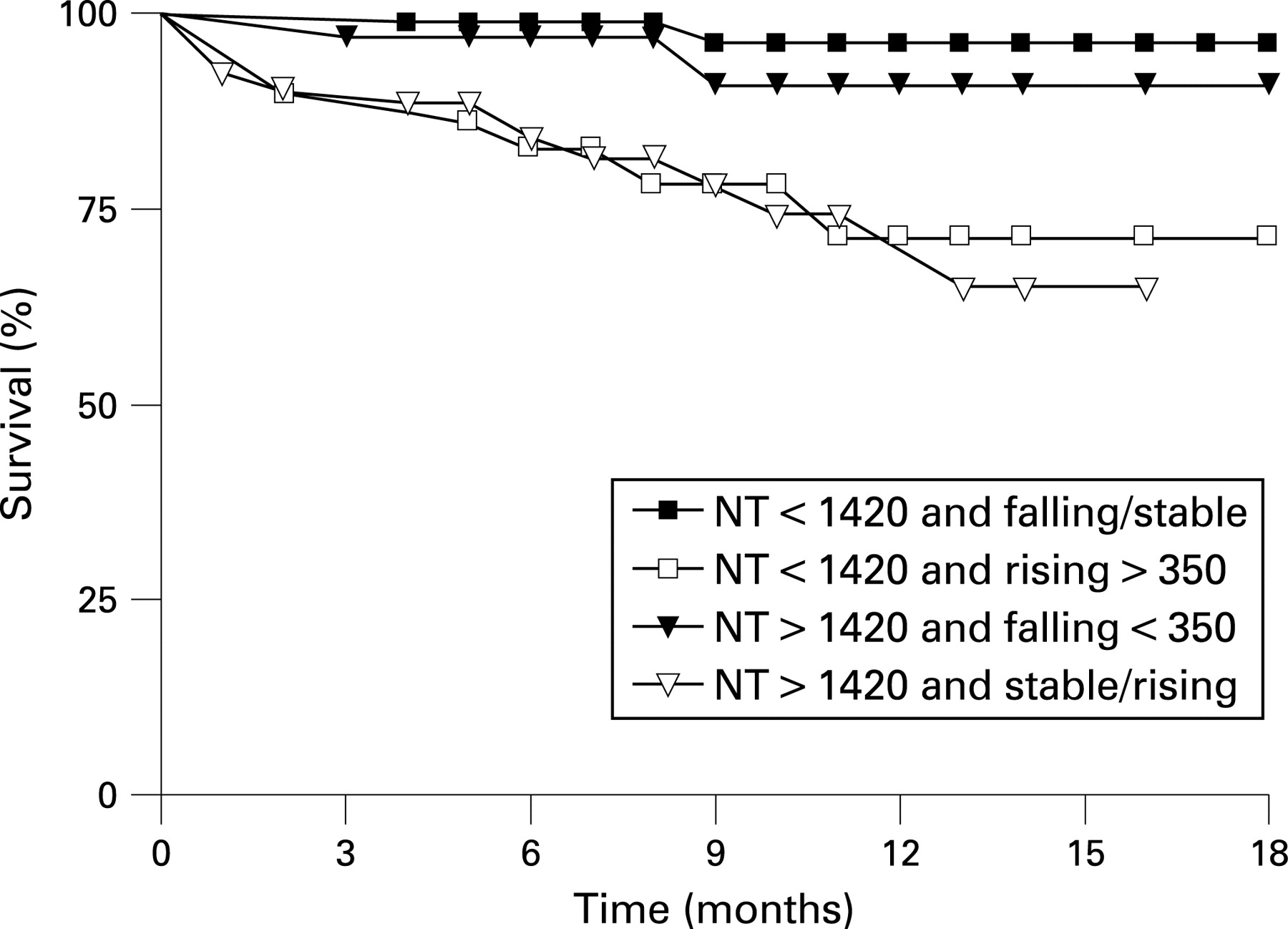
S97 SERIAL NT-PROBNP MEASUREMENTS PREDICT EARLY CLINICAL WORSENING AND MORTALITY IN PULMONARY ARTERIAL AND CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION
C Carlin, KG Blyth, LER McLure, R Spooner, AJ Peacock, MK Johnson. Scottish Pulmonary Vascular Unit, Glasgow, UK
Background: Single measurements of serum B-type natriuretic peptides (BNP and NT-proBNP) have been shown to correlate with clinical and haemodynamic indices and predict prognosis in patients with pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH). We sought to determine the utility of serial measurement of serum [BNP] and [NT-proBNP] in treated PAH and CTEPH patients.
Methods: In the period April 2006–April 2007, 125 patients attending with diagnoses of PAH or CTEPH had serial measurement of serum [BNP] and [NT-proBNP], at 3-month intervals or more, with simultaneous clinical review and 6-minute walk tests. Patients were subsequently followed for a further 6 months and clinical outcomes in relation to the preceding BNP measurements were assessed by receiver operator curve (ROC) and survival curve analysis.
Results: An initial NT-proBNP of less than 1420 pg/ml and/or a fall in NT-proBNP of 350 pg/ml, after 3 months, excluded mortality in the following 6 months (area under ROC curve 0.87, 100% sensitivity). Patients with a baseline NT-proBNP of more than 1420 pg/ml, which did not fall by more than 350 pg/ml, or patients whose NT-proBNP rose by more than 350 pg/ml from baseline had impaired survival in the following months (fig, p<0.001). Results for clinical worsening (right heart failure and/or need for escalation of targeted therapy) at 3 months were comparable. Results with serial [BNP] were similar but serial 6-minute walk distance showed no correlation with future clinical outcome.
Conclusion: Serial NT-proBNP measurement can predict clinical outcomes and should be considered for routine clinical use in pulmonary hypertension practice. [NT-proBNP] could be utilised in a prospective trial of goal-directed therapy for PAH and CTEPH.

{kind=link}
{kind=link}