Article Text

Statistics from Altmetric.com
S81 DEVELOPING A NON-INVASIVE TEST FOR PSEUDOMONAS DETECTION: TOWARDS AN ELECTRONIC NOSE
1S Savelev, 1A De Soyza, 2A Nicholson, 2J Perry, 1M Petrie, 1R Taylor, 3SJ Bourke, 1AJ Fisher, 1PA Corris. 1Institute for Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK, 2Dept Medical Microbiology, Freeman Hospital, Newcastle University, Newcastle upon Tyne, UK, 3Cystic Fibrosis Unit, Royal Victoria Infirmary, Newcastle upon Tyne, UK
Background: Pseudomonas aeruginosa (PA) is a key respiratory pathogen with a distinctive odour in culture. Volatile organic compounds (VOC) are a likely cause of this odour and have previously been detected in the headspace gases of bacterial cultures. Diagnostic improvements are required for the early detection of Pseudomonas infection in at-risk groups such as cystic fibrosis. Limitations in sputum collection (eg, children unable to expectorate) and culture (overgrowth by other bacteria) mean prompt PA eradication therapy may be denied to patients.
Hypothesis: Determining VOC in headspace gases, sputum and exhaled breath may offer benefits in addition to standard culture.
Methods: Solid phase micro-extraction gas chromatography mass spectrometry (SPME–GCMS) using fibres was used to collect headspace gas volatiles initially from overnight bacterial cultures of known cystic fibrosis pathogens or PA. 40 sputum samples were collected from cystic fibrosis and non-cystic fibrosis bronchiectasis clinics and headspace gas analysis after 15 minutes at 37° was compared with 24–48-h standard microbiology culture results. Once potential markers were identified, 10 patients underwent exhaled breath testing using a modified spacer device containing a SPME fibre.
Results: In vitro cultures; azridine was noted in 5/6 PA strains but none of the other bacteria tested (n = 6 including Staphylococcus aureus, Pseudomonas putida and Escherichia coli). In 40 sputum specimens culture revealed PA in 28 and the absence of PA in 12. The headspace gases analysis of the corresponding sputum (in vivo) revealed that azridine was a poor marker of PA, with a sensitivity of 35% and specificity of 60%. Other VOC appeared better biomarkers of PA in sputum headspace, eg, 2 nonanone had sensitivity of 85% and specificity of 95%. Of 15 possible other biomarker VOC a range of sensitivity/specificity levels was noted. In the initial five breath tests studied 2 nonanone was, however, not readily detected.
Conclusions: Volatile organic compounds appear to be released from P aeruginosa dependent on the sample source. In sputum 2 nonanone is a good marker of PA and could add to the speed of detection; it was not, however, readily detectable in exhaled breath. Additional breath testing data are currently being studied.
S82 THE CULTURE OF PRIMARY BRONCHIAL EPITHELIAL CELLS FROM THE LUNGS OF PEOPLE WITH CYSTIC FIBROSIS REMOVED AT THE TIME OF TRANSPLANTATION: A MODEL TO STUDY CYSTIC FIBROSIS LUNG DISEASE
1M Brodlie, 2MC McKean, 3J Perry, 3A Nicholson, 3GE Johnson, 1JP Pearson, 1A Fisher, 1PA Corris, 1JL Lordan, 1C Ward. 1Newcastle University, Newcastle upon Tyne, UK, 2Royal Victoria Infirmary, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK, 3Freeman Hospital, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK
Introduction: Mortality and morbidity in cystic fibrosis (CF) are largely due to lung disease. Despite improvements in survival the exact pathogenesis of CF lung disease remains poorly understood. Ultimately it results in progressive bronchiectasis and premature death. Lung transplantation is the only life-sustaining option for end-stage disease. Studies involving animal models and immortalised cell lines have contributed significantly to our current knowledge of CF lung disease. However, there are inherent limitations to both approaches, including poor replication of lung pathology and failure to reflect in vivo findings. The opportunity to work on primary tissue from patients with CF is rare. A successful ex vivo culture system for primary bronchial epithelial cells (PBEC) from people with CF would therefore represent a valuable cellular model for the study of CF lung disease.
Objective: To culture PBEC from the lungs of people with CF removed at the time of transplantation.
Methods: Pieces of segmental bronchus were removed from lungs immediately after explantation. The tissue was treated with patient-specific antimicrobials and mucolytics to achieve washing and disinfection prior to harvesting of PBEC. Submerged PBEC cultures were established in cells weaned from antimicrobials, before transfer to an air–liquid interface (ALI). Cultured PBEC were characterised by their morphology under light and scanning electron microscopy and pan-cytokeratin and haematoxylin and eosin staining. Culture supernatants and cells were assessed for mucus production by enzyme-linked immunosorbent assay and amylase-periodic acid Schiff staining, respectively.
Results: PBEC have been successfully cultured from nine of 14 patients attempted. Mucus production and tight junction formation has been demonstrated at ALI. The PBEC remain viable after storage in liquid nitrogen. PBEC cultures failed from one patient due to immediate overgrowth with Burkholderia cepacia complex and in four patients initially successful cultures overgrew with Pseudomonas aeruginosa once antimicrobials were withdrawn.
Conclusions: PBEC culture is possible from lungs removed at the time of transplantation from people with CF. Tailored antimicrobial strategies are practicable and yield a favourable success rate. This technique represents a valuable resource that provides a model to elucidate the pathogenic mechanisms in CF lung disease and to investigate potential therapeutic targets.
S83 EFFECT OF PAEDIATRIC INFECTION CONTROL MEASURES IN CYSTIC FIBROSIS: THE ADULT CLINIC’S VIEW
VS Sreeguru Lakshman, E Othman, H Miller, P Dyce, MJ Ledson, MJ Walshaw. Liverpool Heart and Chest Hospital, Liverpool, UK
Introduction: The major cause of death in cystic fibrosis (CF) is respiratory failure, due to a progressive diminution in lung function precipitated by chronic airway infection, in particular with Pseudomonas aeruginosa (PA). The prevention of chronic PA infection, including segregation to prevent the spread of transmissible strains, is an important strategy to improve survival. We were interested to assess whether such measures carried out in our local paediatric clinics had influenced chronic infection rates and hence lung function over time.
Methods: Patients with Burkholderia complex were excluded from the study. We audited the case records of all remaining 160 CF patients (89 males) transferred from paediatric to adult care over the last 10 years (1998–2007), paying attention to lung function, chronic infection status and the prevalence of PA transmissible strains at transition. Chronic infection was defined as three or more positive respiratory samples over a one-year period.
Results: In 1998, all 11 transfers were chronically infected with PA, but this diminished progressively (r = 0.24, p<0.004) such that by 2007, nine transfers (41% of the total) were free of PA infection. Similarly, the proportion infected by transmissible PA strains also progressively fell (r = 0.41, p<0.001), from 72% of the total (eight patients) in 1998 to 36% of the total (eight patients) in 2007. As regards pulmonary function, there was a year-on-year cohort improvement in FEV1 (r = 0.48, p<0.001): comparison between PA infected and the remainder was not possible due to low patient numbers of the latter in the early years.
Conclusion: These results show an improvement in the PA infection status and lung function in CF patients transferred from the paediatric sector over the last 10 years. Moreover, the prevalence of transmissible strains has also reduced, suggesting that patient segregation is an effective way of preventing the spread of these difficult organisms in the CF community.
S84 LARGE ARTERY HAEMODYNAMICS IN CYSTIC FIBROSIS
1JH Hull, 1R Garrod, 2TB Ho, 2RK Knight, 3JR Cockcroft, 3DJ Shale, 3CE Bolton. 1St George’s and Kingston University, London, UK, 2Frimley Park Hospital, Frimley, UK, 3Cardiff University, Cardiff, UK
Introduction and Objectives: Adult cystic fibrosis (CF) is characterised by persistent systemic inflammation and a high prevalence of diabetes mellitus, both of which are now recognised to be important factors in promoting premature vascular aging, as indicated by increased large arterial stiffness. Increased arterial stiffness has important physiological implications for central blood pressure, myocardial work and perfusion and is an independent risk factor for the development of cardiovascular disease. We hypothesised that arterial stiffness would be increased in adults with CF and would relate to inflammation and the presence of diabetes.
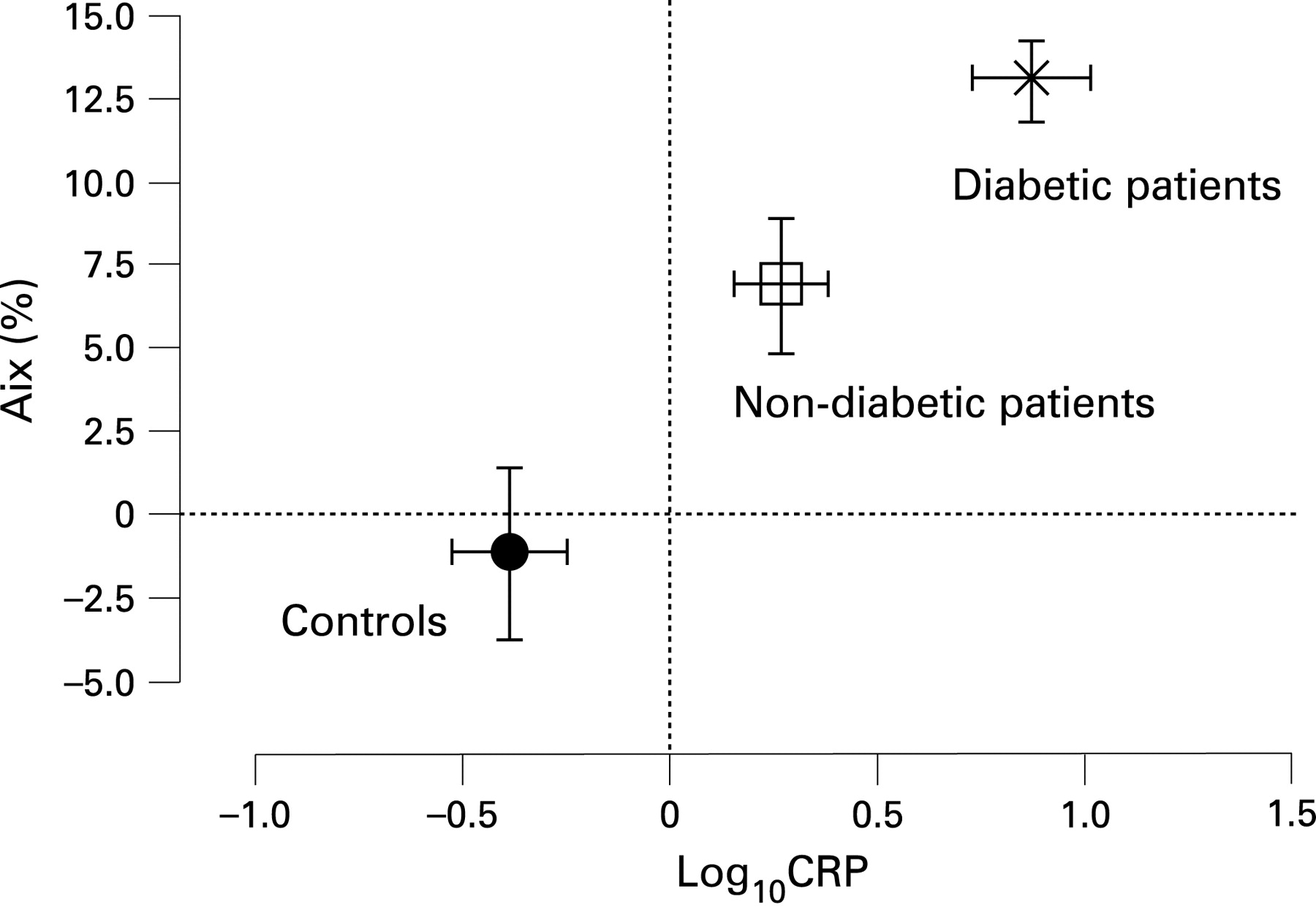
Methods: We studied 50 (33 male) adults with stable CF, mean (SD) age 28.0 years (8.2), body mass index (BMI) 22.2 kg/m2 (3.1), FEV1 % predicted 65.2% (21.2) and 26 age/gender/BMI-matched healthy controls. Arterial stiffness was assessed by applanation tonometry to obtain augmentation index (AIx) (SphygmoCor), the preferred measure of arterial stiffness in this age group and aortic pulse wave velocity (APWV). In addition we measured blood pressure, spirometry, glucose tolerance status (in non-diabetics), physical activity, serum electrolytes, C-reactive protein (CRP), lipid profile and calculated the glomerular filtration rate.
Results: Height, mean arterial pressure, physical activity were similar between patients and controls but CRP was greater (p<0.005). AIx was greater in patients, 8.5% (11.1) than controls −1.8% (13.1) and was greatest in diabetic patients (n = 13), 13.1% (4.3) (analysis of variance, p<0.05). Importantly, AIx remained greater in non-diabetic patients, 6.9% (12.3) than controls (fig). APWV was similar in patients, 6.1 m/s (0.8) and controls, 6.4 m/s (1.5). In patients, AIx was directly related to age (r = 0.54, p<0.001) and log10 CRP (r = 0.33, p<0.05) and inversely with FVC % predicted (r = −0.34, p<0.05). In addition, AIx was inversely related to glomerular filtration rate (r = −0.55, p<0.001) but did not relate to lipid profile.
Conclusions: Adult patients with CF have evidence of premature vascular aging, as indicated by increased AIx compared with controls. This relates to systemic inflammatory mediators and diabetic status.

{kind=link}
S85 PREDICTORS OF OUTCOME IN CYSTIC FIBROSIS PATIENTS ADMITTED TO INTENSIVE CARE
JF Greenwood, VS Sreeguru Lakshman, H Miller, C Cowperthwaite, MJ Ledson, MJ Walshaw. Liverpool Heart and Chest Hospital, Liverpool, UK
Introduction: The decision to admit adult patients with cystic fibrosis (CF) to the intensive care unit (ICU) is controversial because of a perceived poor outcome and the likely need for prolonged multiorgan support. Although scoring systems (APACHE II, SAPS II, MODS and SOFA) are available to help predict morbidity and mortality following ICU admission for the general population, their relevance to CF patients is unknown. To look at this further, we looked at the performance of these scoring systems in patients attending our large adult CF unit.
Patients and Methods: All 10 CF patients (mean age 21 years (range 17–27), seven respiratory failures, two upper gut haemorrhages, one following thoracotomy, four male) admitted to ICU over 5 years (2002–7) formed the study population. Data on chronic disease history and acute physiological parameters were collected and scores were calculated from the ICU admission dataset.
Results: Mean ventilated days was 5 (0–12), mean ICU stay was 5.6 days (1–14) and ICU mortality was 60%. Only two patients were alive at one year. Low body weight was associated with mortality (p = 0.02), but the presenting complaint, spirometry, recent hospital admission, presence of diabetes, transplant listing or admission blood gases did not predict outcome. In terms of scoring systems, only SAPS II was significantly associated with mortality both on ICU (p = 0.029) and at one year (p = 0.04). Both MODS and SOFA showed a trend towards predicting outcome, but APACHE II was poor at predicting both ICU and one-year mortality in our patients.
Conclusion: CF patients admitted to ICU have a poor outcome, both immediately and within one year. With the exception of SAPS II, organ dysfunction scores used for the general population are not useful in predicting outcome in these patients. The association of low body weight with mortality again highlights the careful attention that needs to be paid to the nutritional status of CF patients.