Article Text

Statistics from Altmetric.com
S58 MECHANICAL STIMULATION OF HUMAN LUNG MICROVASCULAR ENDOTHELIAL CELLS CAUSES REMODELLING OF THE ALVEOLAR-CAPILLARY MEMBRANE AND DYSPNOEA IN CHRONIC HEART FAILURE
1JES Park, 2AR Lyon, 2SE Harding, 1MJD Griffiths. 1Unit of Critical Care, NHLI, Imperial College, London, UK, 2Department of Cardiac Medicine, NHLI, Imperial College, London, UK
Introduction: Dyspnoea is a common and debilitating symptom of chronic heart failure (CHF). Patients with CHF have reduced pulmonary microvascular permeability and gas transfer that persists after effective treatment of pulmonary oedema, suggesting a structural abnormality of the alveolar-capillary membrane (ACM). Structural remodelling of the lungs in CHF is characterised by proliferation of myofibroblasts with excess matrix deposition in alveolar septae. Endothelin-1 (ET-1), a potent vasoconstrictor and mitogen, binding endothelin A receptor and endothelin B receptor on human fibroblasts stimulates collagen 1 synthesis. The plasma ET-1 concentration in CHF correlates with the degree of haemodynamic disturbance, dyspnoea and mortality. We propose that in CHF, pulmonary venous hypertension and resultant mechanical strain on the pulmonary microvasculature stimulates the release by endothelial cells of “fibrogenic” mediators including ET-1. This process contributes to ACM remodelling and dyspnoea in patients.
Methods: The expression of message for components of the ET-1 pathway was investigated in strained (static mechanical strain 20% elongation for 4 h, Flexercell 4000) monolayers of human lung microvascular endothelial cells (HLMVEC; Lonza, UK) and in whole lungs of rats with or without heart failure 16 weeks after myocardial infarction (MI) or a sham procedure.1 Supernatants from HLMVEC strained in the presence phosphoramidon (0–5 mmol), an endothelial converting enzyme inhibitor were added to serum starved human fetal lung fibroblasts (HFL-1) to assess cellular proliferation (CyQUANT assay).
Results: Expression of ppET-1 by HLMVEC increased after SMS and in whole lungs of rats after MI, with a decrease in endothelin B receptor message also seen in lungs post-MI (fig). Supernatants from HLMVEC stretched in the absence but not in the presence of phosphoramidon (5 mmol), which abolished stretch-induced ET-1 release, increased proliferation by HFL-1 cells.
Conclusion: Mechanical strain stimulates the release of ET-1 by HLMVEC, which may cause local fibroblast proliferation. An increase in ET-1 message is also seen in whole rat lungs at 16 weeks post-MI.
Funding: JESP is supported by a MRC clinical research training fellowship.

S59 THE OSTEOPROTEGERIN–TNF-RELATED APOPTOSIS-INDUCING LIGAND AXIS IN PULMONARY ARTERIAL HYPERTENSION
1A Lawrie, 1C Paiva, 2M Southwood, 2J Suntharalingam, 1SE Francis, 1DC Crossman, 2NW Morrell, 1CMH Newman. 1University of Sheffield, Sheffield, South Yorkshire, UK, 2University of Cambridge, Cambridge, Cambridgeshire, UK
Pulmonary arterial hypertension (PAH) is a devastating disease characterised by the narrowing and occlusion of small pulmonary arteries due to intimal hyperplasia. We have recently reported that osteoprotegerin, a member of the tumour necrosis factor (TNF) superfamily, is upregulated in serum and lesions of patients with idiopathic PAH. We have also shown that signals from BMP-R2, 5-HT and IL-1 converge upon and upregulate osteoprotegerin expression and that osteoprotegerin induces both proliferation and migration of PA-SMC in vitro, suggesting that osteoprotegerin plays a central role in the pathogenesis of PAH. Interestingly, osteoprotegerin is known to interact with TNF-related apoptosis-inducing ligand (TRAIL) and there is emerging evidence to suggest that TRAIL also plays an important role in vascular tissues. We hypothesised that TRAIL expression is also dysregulated in PAH and that this induces the proliferation and migration of PA-SMC, consistent with a role in the pathogenesis of PAH. TRAIL exists as a type II TNF family transmembrane protein. The extracellular domain can be proteolytically cleaved from the cell surface to act as a soluble cytokine (sTRAIL) capable of binding to five TRAIL receptors, (TRAIL-R1–4) and osteoprotegerin. We found that TRAIL expression was increased in lesions from patients with idiopathic PAH, but that the circulating levels of sTRAIL were significantly lower in idiopathic PAH patient serum compared with controls, leading to a significant difference in the osteoprotegerin : TRAIL ratio between the groups. In vitro, recombinant TRAIL was found to induce both proliferation and migration of human PA-SMC in a dose-dependent manner, a response requiring the phosphorylation of extracellular signal regulated kinase 1/2. Interestingly, the combination of the optimal concentrations of osteoprotegerin and TRAIL prevented both proliferation and migration in PA-SMC, suggesting that the relative concentration of these molecules is important in terms of cell phenotype. Although further work is clearly required, including the study in other groups of PAH patients, these data suggest that the combined increase in circulating osteoprotegerin and the decrease in circulating sTRAIL may play an important role in disease pathogenesis. The ratio of osteoprotegerin : TRAIL may provide a new biomarker for tracking disease progression.
S60 LYMPHOCYTE PROFILES IN IDIOPATHIC AND CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION
E Soon, M Southwood, M Toshner, K Sheares, J Pepke-Zaba, AR Exley, NW Morrell. Papworth Hospital NHS Trust, Cambridge, UK
Introduction: Autoimmunity is thought to contribute to the pathobiology of pulmonary arterial hypertension (PAH), although the mechanisms remain unclear. Evidence in support of immune mechanisms include the high prevalence of antinuclear antibodies,1 the strong association with certain autoimmune diseases such as scleroderma,2 and evidence from animal models.3 We hypothesised that patients with idiopathic PAH may demonstrate differences in lymphocyte profiles compared with normal or disease controls.
Methods: Flow cytometry was used to assess the lymphocyte subsets in patients with idiopathic PAH (n = 10), heritable PAH with bone morphogenetic protein receptor-2 (BMPR2) mutations (n = 3), chronic thromboembolic pulmonary hypertension (CTEPH; n = 8) and healthy age and sex-matched controls (n = 8). All cases were clinically stable and patients with known autoimmune disease, ongoing infection or malignancy were excluded. In addition, explanted lung tissue from PAH patients (n = 20) undergoing transplantation were immunostained for lymphocyte markers to immunophenotype lymphocyte subsets.
Results: These are summarised in the table. Values are expressed as percentages of circulating cells. The percentage of circulating CD4+CD25+ cells was higher in idiopathic PAH but did not quite reach significance (p = 0.07). When this was narrowed down to the T-regulatory subset (CD4+CD25+CD127−) the BMPR-2 patients displayed a significant increase. There was also a significant decrease in CD19+ B cells in the idiopathic PAH patients. There was no difference in circulating percentages of CD3+ lymphocytes, CD3+CD4+ T helper cells, CD3+CD8+ cytotoxic T cells or CD56+ natural killer cells. Immunohistochemical staining has also demonstrated the presence of frequent CD8+ cells and occasional T-regulatory cells surrounding the vascular lesions associated with PAH.
Conclusions: The T-regulatory subset is increased in patients with heritable PAH with BMPR-2 mutations. This is not demonstrated in CTEPH. Further investigation is required to see if this is a pathological or a compensatory mechanism.
S61 CHARACTERISATION OF PULMONARY VASCULAR REMODELLING IN A MOUSE MODEL OF SCHISTOSOMIASIS
A Crosby, F Jones, M Southwood, D Dunne, N Morrell. Cambridge University, Cambridge, UK
Introduction: Schistosomiasis is considered to be the commonest worldwide cause of pulmonary hypertension. At present there is no established animal model to study the pathobiology of this important condition. We sought to develop and characterise a mouse model that recapitulates the pulmonary vascular pathology seen in patients with schistosomiasis.
Methods: Mice (C57/BL6) were infected percutaneously with a high dose (∼75–100 cercariae) or low dose (∼30 cercariae) of Schistosoma mansoni and the development of lung and liver pathology was studied in the subacute (high dose) and chronic (low dose) settings. We determined the degree of right ventricular hypertrophy from the ratio of the weight of the right ventricle to that of the left ventricle plus septum. Liver and lung egg counts were performed at specified time points. Pulmonary vascular remodelling was assessed by morphometry following immunohistochemical staining with antibodies for smooth muscle alpha actin and the endothelial marker, von Willebrand factor.
Results: In the subacute, high dose setting mice showed few eggs in the lungs and no evidence of pulmonary vascular remodelling. In contrast, chronically infected animals had a much greater lung egg burden and developed marked pulmonary vascular remodelling, accompanied by perivascular inflammation from 12 weeks onwards. In addition, we observed the presence of plexiform-like lesions in these mice. Lung egg burden positively correlated with both liver egg burden and the degree of right ventricular hypertrophy in the chronic group.
Conclusions: This study provides evidence for extensive pulmonary vascular remodelling in a mouse model of pulmonary schistosomiasis, including the formation of plexiform-like lesions. The degree of remodelling and right ventricular hypertrophy was related to the lung egg burden. This experimental model will be of use to study the pathobiology and treatment of pulmonary hypertension associated with schistosomiasis.
S62 PULMONARY ARTERY FIBROBLASTS DRIVE SMOOTH MUSCLE CELL PROLIFERATION IN A PULMONARY HYPERTENSIVE RAT MODEL
AC Church, AJ Peacock, D Welsh. Scottish Pulmonary Vascular Unit, Glasgow, UK
Introduction: The cause of cellular proliferation leading to vascular remodelling in pulmonary arterial hypertension (PAH) is unknown. We have shown that exposure of pulmonary artery fibroblasts (PAF) to acute hypoxia releases a diffusible factor(s), which causes proliferation of pulmonary artery smooth muscle cells (PASMC) via a p38MAPkinase (MAPK)-dependent mechanism. We set out to determine if these same mechanisms hold true in a chronic hypoxic pulmonary hypertensive rat model in which pulmonary vascular remodelling is established.
Methods: Primary cell cultures of PAF and PASMC were isolated from rats exposed to hypobaric (550 mBar) hypoxia for 14 days (PHrat) and controls (Nrat) at atmospheric pressure. PAF from both controls and PHrat were treated with low dose fetal calf serum (0.1–0.5%) for 24 h and the supernatant was then transferred to quiescent PASMC in 24-well plates for a further 24 h. All experiments were performed in normoxia. [3H]-Thymidine assays were used to determine cellular proliferation. Total and phospho-p38MAPK levels in the PAF were assessed by Western blotting. The effect of blocking p38MAPK on the release of mitogenic factors was investigated by incubating the PAF with SB-205380, a specific p38MAPK inhibitor.

Results: There was no proliferative response of the SMC to the low dose serum applied directly to the PASMC (mean cpm 21.5; SD ± 3.41) (see fig). Although there was some proliferation of PASMC from the supernatant of Nrat PAF, there was a twofold greater proliferation observed with the addition of conditioned media across all ranges of serum from the PHrat PAF (74.4 SD ± 9.4 vs mean cpm 158.6 SD ± 11.4; p<0.005). The addition of media obtained from PAF incubated with SB203580 to PASMC abrogated the increased proliferation (data not shown). Western blot confirmed that active phospho-p38 MAPK was increased in the 0.1% serum stimulated cells and absent in the SB203580 treated cells. Total p38 MAPK was the same in all conditions.
Conclusions: We have shown that fibroblasts from a PAH animal model release yet unidentified mitogenic factor(s) in normoxic conditions, which drives the proliferation of PASMC and is dependent on p38MAPK. Fibroblasts may play an important role in the remodelling process of PAH by inducing proliferation of other pulmonary vascular cell types.
S63 DEXAMETHASONE IMPROVES HAEMODYNAMICS IN ESTABLISHED INFLAMMATORY PULMONARY ARTERIAL HYPERTENSION IN A RAT MODEL
1LC Price, 1D Montani, 1F Perros, 1C Tcherakian, 2LS Howard, 1G Simonneau, 1M Humbert. 1Hopital Antoine Beclere, Clamart, Paris, France, 2The Hammersmith Hospital, London, UK
Introduction and Objectives: There is increasing evidence for the contribution of inflammation to pulmonary vascular remodelling in pulmonary arterial hypertension (PAH), but little on the role of anti-inflammatory therapies in its treatment. Our group and others have shown that dexamethasone can prevent PAH in rats when co-administered with monocrotaline to induce PAH. Monocrotaline is known to induce PAH by 2 weeks and we wished to test the hypothesis that dexamethasone would reverse established PAH at this stage, making it more relevant to human disease.
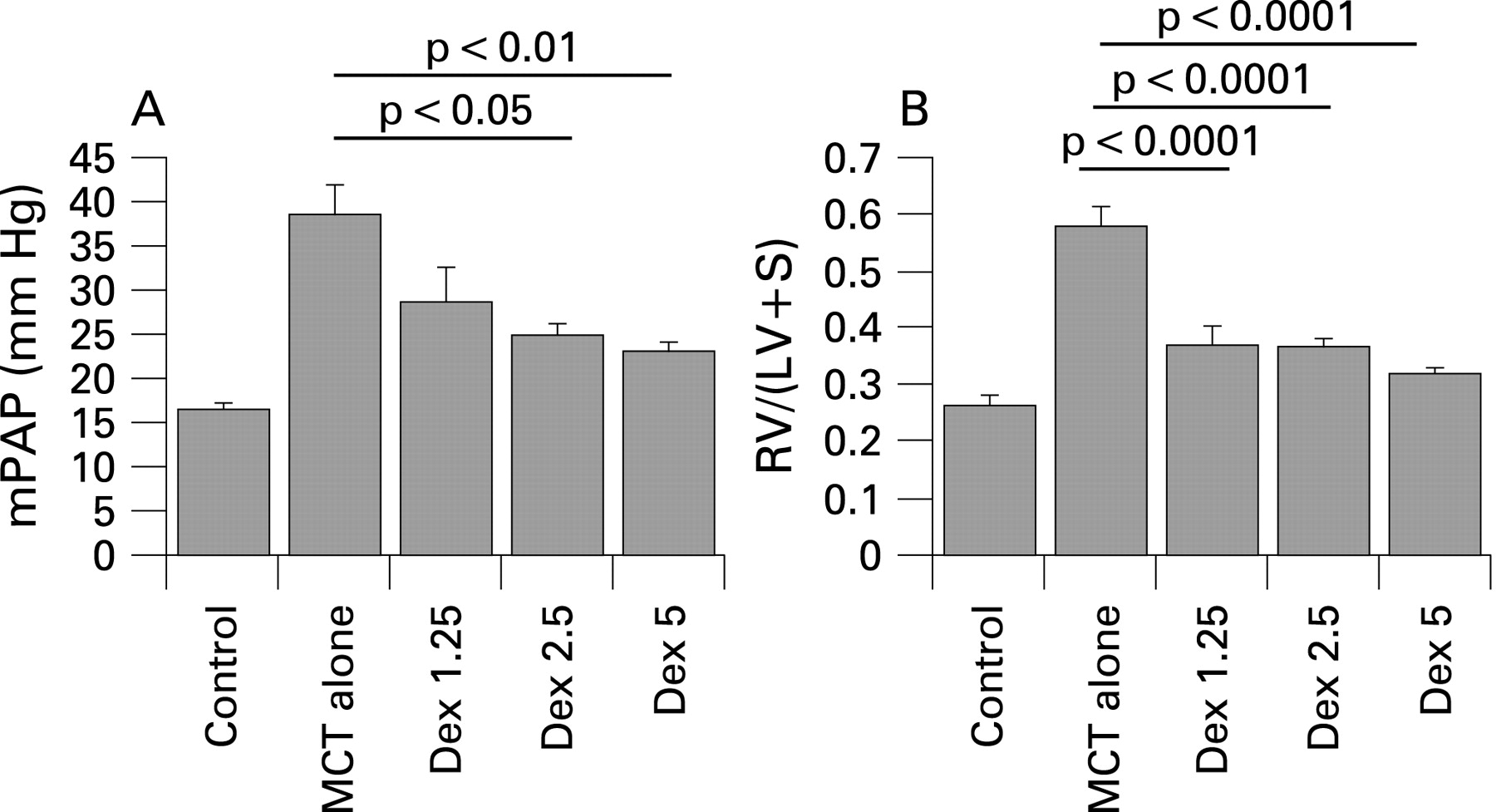
Methods: Male wistar rats (100 g) were divided into five groups: controls receiving no monocrotaline, those receiving monocrotaline alone and those receiving monocrotaline then dexamethasone 1.25 mg/kg or 2.5 mg/kg alternate days (Dex1.25, Dex2.5) or 5 mg/kg daily (Dex5). Monocrotaline (60 mg/kg subcutaneous injection) was administered on day 1 and dexamethasone was given by intraperitoneal injection from days 15 to 28. At day 28, all rats were anaesthetised (35 mg/kg ketamine, 4 mg/kg xylasine and 0.5 mg/kg acepromazine) and haemodynamics measured using a 3.5 French umbilical vessel catheter. The ratio of right to left ventricular and septal weight was recorded (RV/LV+S).
{kind=link}
{kind=link}
{kind=link}
Results: At day 28, the monocrotaline-alone rats had severe PAH with mean pulmonary arterial pressure (mPAP) (40.8 ± 6.4 mm Hg vs 16.4 ± 1.5 mm Hg, p<0.001), right ventricular systolic pressure (RVSP) (94.3 ± 7.8 mm Hg vs 35.2 ± 2.4, p<0.001) and RV/LV+S (0.6 ± 0.1 vs 0.26 ± 0.06, p<0.001) compared with controls. PAH was reversed in all the groups treated with dexamethasone with a suggestion of a dose–response effect, with mPAP falling to 28.7 ± 11.9 (p = 0.07 compared with monocrotaline alone), 24.9 ± 4.7 (p<0.05) and 23.1 ± 3.5 (p<0.01) in Dex1.25, Dex2.5 and Dex5, respectively (fig A). RVSP was also significantly lower in all three groups. Right ventricular hypertrophy as assessed by RV/LV+S was also reversed by dexamethasone compared with monocrotaline alone (p<0.001 in all groups, fig B). None of the measurements in the Dex5 group were statistically different to controls. There was a significant improvement in survival between all the dexamethasone groups when compared with monocrotaline alone (log rank test p<0.001).
Conclusions: These results suggest that monocrotaline-induced PAH can be reversed by steroids, raising the possibility that an anti-inflammatory strategy may be beneficial in some cases of pulmonary hypertension.