Article Text

Statistics from Altmetric.com
T1 MESENCHYMAL STEM CELLS REDUCE LUNG METASTASES BY CONTROLLED TARGETED DELIVERY OF TUMOUR NECROSIS FACTOR-RELATED APOPTOSIS-INDUCING LIGAND
MR Loebinger, SM Janes. Centre for Respiratory Research, University College London, London, UK
Background: Recent studies suggest that bone marrow-derived mesenchymal stem cells (MSC) incorporate within tumour stroma. We hypothesised that MSC engineered to produce and deliver tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), a transmembrane protein that causes selective apoptosis of tumour cells, would home to and kill cancer cells in a lung metastatic cancer model.
Methods: TRAIL and the IRES-eGFP reporter gene, under the control of a tetracycline promoter were generated and subcloned into an HIV-1-based self-inactivating lentiviral vector. Expression of GFP and TRAIL in vitro, in 293T cells and MSC (MOI 10) was confirmed by flow cytometry, reverse transcription PCR, Western blot and ELISA. Apoptotic function of MSC expressing TRAIL (GFP positive) was established in co-culture with cancer cell lines (Hela, MDAMB231 and A549) (DiI-labelled) using Annexin V flow cytometry and microscopy. MSC homing was examined using chemotaxis assays with or without chemokine blocking antibodies. In vivo subcutaneous and systemically delivered metastatic lung xenograft cancer models were performed in NOD/SCID mice (MDAB231 cells).
Results: MSC were transduced with greater than 80% efficiency (flow cytometry) and expressed TRAIL under the control of doxycycline (reverse transcription PCR, ELISA, Western blot). Transduced and activated MSC caused lung (A549), breast (MDAB231) and cervical (Hela) tumour cell death and apoptosis in co-culture experiments (all p<0.001). Tumour cell death was maintained at low (1 : 16) MSC/tumour cell ratios (p<0.001) and reduced by a TRAIL neutralising antibody (p<0.001). Tumour cells expressing a dominant negative Fas-associated death domain (dnFADD) showed that TRAIL induced death was via the extrinsic apoptotic pathway (p<0.001). Transwell migration studies demonstrated preferential homing of MSC to tumour cells compared with control cells (p<0.001), which was not significantly blocked by anti-CXCL12 antibody. Subcutaneous xenograft experiments demonstrated that directly delivered TRAIL-expressing MSC reduced tumour growth (p<0.001). Intravenously injected DiI-labelled MSC demonstrated homing and engraftment into lung metastases. TRAIL-expressing MSC delivered to doxycycline-treated mice with lung metastases caused a significant reduction in lung weight (p<0.01) and metastasis number (p<0.001) compared with controls. Metastastic disease was cleared completely in 3/8 MSC/doxycycline-treated mice compared with 0/18 controls (p = 0.052).
Conclusion: This is the first study to demonstrate a significant reduction in tumour burden with inducible TRAIL-expressing MSC.
T2 ADRENERGIC β2-RECEPTOR GENOTYPE PREDISPOSES TO EXACERBATIONS IN ASTHMATIC PATIENTS ON AS-REQUIRED SHORT-ACTING AND REGULAR LONG-ACTING β2 AGONISTS
1K Basu, 2CNA Palmer, 2R Tavendale, 3BJ Lipworth, 4S Mukhopadhyay. 1Maternal and Child Health Sciences, Ninewells Hospital and Medical School, University of Dundee, Dundee, UK, 2Population Pharmacogenetics Group, Biomedical Research Centre, University of Dundee, Dundee, UK, 3Asthma and Allergy Research Group, Division of Medicine and Therapeutics, Ninewells Hospital, Dundee, UK, 4Royal Alexandra Children’s Hospital, Brighton and Sussex Medical School, Brighton, UK
Introduction and Objectives: Inhaled short-acting β2-agonists used on an “on demand” basis constitute one of the most commonly prescribed asthma medications in the world. Earlier, in a smaller number of participants (n = 546), we have shown that the arginine-16 (Arg16) genotype of ADRB2 predisposes to exacerbations in young asthmatic patients on regular salmeterol. Here we explored the role of Arg/Arg16 status on asthma exacerbations in the context of the use of “on demand” salbutamol in a larger population. In addition, we sought to replicate the previously observed effects of this genotype on a larger population of asthmatic patients on regular salmeterol.
Methods: A cross-sectional survey was undertaken using electronic records, direct interviews and genotype determination at position 16 of ADRB2 in DNA from mouthwash samples of 1161 asthmatic children and young adults in Scotland during 2004–7. Asthma exacerbations, medication use over 6 months and lung function were studied.
Results: There was a significant increase in overall exacerbations, in asthmatic patients with Arg/Arg compared with Gly/Gly or Arg/Gly, in the absence of inhaled long-acting β-agonist therapy (Arg/Arg 49%, Arg/Gly 38%, Gly/Gly 36%; p = 0.016, odds ratio (OR) 1.85). In addition, we observed a deleterious effect of Arg/Arg status on exacerbations in young asthmatic patients receiving regular salmeterol (Arg/Arg 65%, Arg/Gly 50%, Gly/Gly 39%; p = 0.040, OR 2.85). Arg/Arg patients had a greater risk of overall exacerbations (OR 1.80, 95% CI 1.24 to 2.62; p = 0.002), were more likely to require oral steroids (OR 1.78, 95% CI 1.17 to 2.70; p = 0.007) and experience school absences (OR 1.76, 95% CI 1.21 to 2.57; p = 0.003) in comparison with Arg/Gly or Gly/Gly variants.
Conclusion: Our data show that the Arg16 homozygous genotype of ADRB2 predisposes to exacerbations in asthmatic children and young adults receiving “on demand” inhaled salbutamol. In addition, we have re-confirmed our previous observations of an adverse role for Arg 16 homozygous status in asthmatic patients on regular salmeterol. This could contribute to the overall asthma burden. There is a need to evaluate the safety and efficacy of inhaled short and long-acting β2-agonists in asthmatic children and young adults homozygous for the Arg/Arg 16 genotype.
T3 AIRWAY SMOOTH MUSCLE HYPERPLASIA IN ASTHMA: A CONSEQUENCE OF FIBROCYTE RECRUITMENT
S Siddiqui, R Saunders, D Kaur, C Doe, A Sutcliffe, F Hollins, P Bradding, A Wardlaw, C Brightling. Institute for Lung Health, Leicester, UK
Background: Airway smooth muscle hyperplasia is a hallmark of asthma. It is associated with disease severity and persistent airflow obstruction. We have considered whether the recruitment of fibrocytes, a population of peripheral blood mesenchymal progenitors, contributes to the development of airway smooth muscle hyperplasia.
Methods: We assessed the number of fibrocytes in bronchial biopsies and peripheral blood from subjects with mild refractory asthma compared with healthy controls. In vitro we investigated the potential mechanisms controlling fibrocyte migration towards the airway smooth muscle bundle.
Results: Fifty-one subjects with asthma and 34 controls were studied. The median (interquartile range) number of fibrocytes/mm2 in the lamina propria was increased in those with refractory disease (1, 9 (1, 7)) compared with mild–moderate asthmatic patients (0 (0.6)), and healthy controls (0 (0)) (p<0.001). The percentage airway smooth muscle area was increased in those with refractory 19.5% (3.6) and mild–moderate asthma 23.3% (1.9) compared with healthy controls 10.5% (1.4) (p = 0.0016, analysis of variance; ANOVA). The number of fibrocytes/mm2 was increased in the airway smooth muscle bundle in subjects with severe refractory asthma (3.8 (9.4)) and mild–moderate asthma (1.1 (2.4)) compared with healthy controls (0 (0)) (p = 0.001). The number of fibrocytes and mast cells within the airway smooth muscle bundle was correlated for the whole group (r = 0.58; p = 0.001) and in asthmatic patients alone (r = 0.44; p = 0.035). In the peripheral blood the fibrocyte number was also increased in severe refractory asthma 1.4 (2.6) compared with healthy controls 0.4 (1.0), but not mild–moderate asthma 1.0 (3.1) × 104/ml (ANOVA p = 0.005, severe-refractory versus healthy controls; p = 0.002). There was no correlation between the number or proportion of peripheral blood/tissue fibrocytes and the subjects’ clinical or physiological characteristics or airway smooth muscle mass. We identified that in vitro airway smooth muscle promotes fibrocyte chemotaxis and chemokinesis (distance of migration after 4.5 h 31 mm (2.9)) versus 17 mm (2.4); n = 20; p = 0.001), which was in part mediated by platelet-derived growth factor (mean (95% CI) inhibition by neutralising antibody 16% (2 to 32); p = 0.03), but not by activation of chemokine receptors.
Conclusions: This study provides the first evidence that the recruitment of fibrocytes to the airway smooth muscle is a feature of asthma and that airway smooth muscle-derived mediators have the capacity to recruit fibrocytes.
T4 CONDITIONAL POSTNATAL DELETION OF TBRII DEFINES A PIVOTAL ROLE FOR RESIDENT FIBROBLASTS IN THE FIBROTIC RESPONSE TO LUNG INJURY
1RK Hoyles, 1K Khan, 1X Shiwen, 2AU Wells, 1DJ Abraham, 1CP Denton. 1Royal Free and University College Medical School, London, UK, 2Royal Brompton Hospital, London, UK
Background: Increased transforming growth factor beta (TGFβ) 1 bioactivity is implicated in fibrosing diseases. Potential sources of matrix in fibrosis include resident fibroblasts, epithelial–mesenchymal transdifferentiation and circulating mesenchymal progenitor cells (fibrocytes). Using fibroblast-specific expression of tamoxifen-dependent Cre recombinase (Cre-ER) we have selectively deleted type II TGFβ receptors (TbRII) postnatally from connective tissue fibroblasts but not other cell types (null). This study explores the effect of disrupting TGFβ signalling in resident fibroblasts on the response to experimental lung injury.
Methods: Adult littermate wild-type (WT) or null mice aged 6–8 weeks were injured with intratracheal bleomycin (WT-B, null-B) or saline (WT-S, null-S). Lung tissue was collected after 7 or 14 days for histological, protein or RNA analysis.
Results: 14 days after injury, WT-B lungs demonstrated a florid fibroproliferative response and strong α-SMA immunoreactivity suggesting activated myofibroblast-like cells. In contrast, little fibrosis or α-SMA signal was seen in null-B. The fibrotic response was quantified in lung homogenates by the Sircol assay, which showed that non-fibrillar cross-linked collagen was significantly attenuated in null-B compared with WT-B at 14 days (23 ± 10 cf 52 ± 4 mg/g wet weight collagen, p = 0.002). Quantitative PCR showed that CTGF, Col1a1 and PAI-1 mRNA expression were significantly attenuated in null-B compared with WT-B (all <0.05). TbRII immunoreactivity was significantly reduced in null-B compared with WT-B (2 ± 1 cells/high power field (hpf) null cf 18 ± 4 cells/hpf WT, p<0.001). Deletion of TβRII in resident fibroblasts had little impact on the degree of alveolar epithelial cell (AEC) II hyperplasia in null animals after injury (WT-B 214 ± 38 TTF-1 positive cells/hpf compared with null-B 198 ± 8 cells/hpf, p = 0.45) and importantly confirmed equivalent AEC injury in this strain. Attenuated fibrosis in null-B was not secondary to reduced circulating active TGFβ-1, which was similar in WT-B and null-B.
Conclusion: We have shown that selective disruption of TGFβ signalling in resident lung fibroblasts protects from experimental lung fibrosis. TbRII is intact in AEC or fibrocytes in the absence of further tamoxifen at the time of lung injury. This suggests that resident fibroblasts are responsible for matrix production in this model.
T5 BMPR-II DYSFUNCTION LEADS TO ABNORMAL GROWTH RESPONSES TO TRANSFORMING GROWTH FACTOR BETA IN PULMONARY ARTERY SMOOTH MUSCLE CELLS: ROLE OF UPREGULATION OF NUCLEAR FACTOR KAPPA B SIGNALLING
1RJ Davies, 1PD Upton, 1L Long, 2R Trembath, 1NW Morrell. 1Division of Respiratory Medicine, University of Cambridge School of Clinical Medicine, Addenbrooke’s and Papworth Hospitals, Cambridge, UK, 2King’s College London, London, UK
Background: Pulmonary arterial hypertension is characterised by increased growth of pulmonary vascular smooth muscle cells. The familial variant of this condition (familial pulmonary arterial hypertension; FPAH) is caused by mutations in the bone morphogenetic protein type II receptor (BMPRII). We have found that pulmonary arterial smooth muscle cells (PASMC) harbouring mutations in BMPR-II are resistant to the growth inhibitory effect of transforming growth factor beta (TGFβ) 1. Here we provide evidence for the mechanism underlying this observation.
Methods: PASMC were harvested from explanted lungs from patients undergoing lung transplantation for FPAH and from heterozygous BMPR-II null mice. We also studied BMPR-II signalling following siRNA knockdown. Cellular proliferation was measured by a trypan blue exclusion method. Activation of BMP, TGFβ-1 and nuclear factor kappa B (NFκB) signalling was examined using immunoblotting, luciferase reporter gene analysis and quantitative reverse transcription PCR.
Results: TGFβ-1 significantly inhibited the growth of human and mouse control cells. In contrast, cells harbouring dysfunctional BMPR-II were resistant to this growth inhibition. Mutant BMPR-II cells displayed reduced Smad 1/5 phosphorylation, reduced transcription of BRE-luciferase reporter as well as reduced expression of BMP target genes (Id1, 2 and 4) in response to BMP4. However, in contrast to previous reports of reciprocal regulation of TGFβ and BMP regulated Smad signalling we found no evidence for increased TGFβ dependent Smad2/3 signaling or gene transcription in mutant cells. A microarray screen of TGFβ-driven gene expression indicated an increase in NFκB-dependent signalling in BMPR-II mutant PASMC. Quantitative reverse transcription PCR confirmed increased baseline expression of the NFκB-dependent genes, IL-6 and IL-8 in mutant PASMC compared with controls. Furthermore, incubation of BMPR-II mutant PASMC with either an IL-6 or an IL-8 neutralising antibody re-sensitised the mutant cells to the growth inhibitory effects of TGFβ.
Conclusions: BMPR-II dysfunction consistently eliminates the growth inhibitory response to TGFβ-1 in PASMC. This effect was independent of Smad signalling. Our data indicate that the abnormal growth response to TGFβ in mutant cells is due to abnormal NFκB signalling and increased secretion of growth-promoting cytokines. This provides an important link between BMPR-II dysfunction and vascular inflammation in FPAH. Inhibition of NFκB signalling and inflammatory cytokines may therefore be a promising therapeutic target in FPAH.
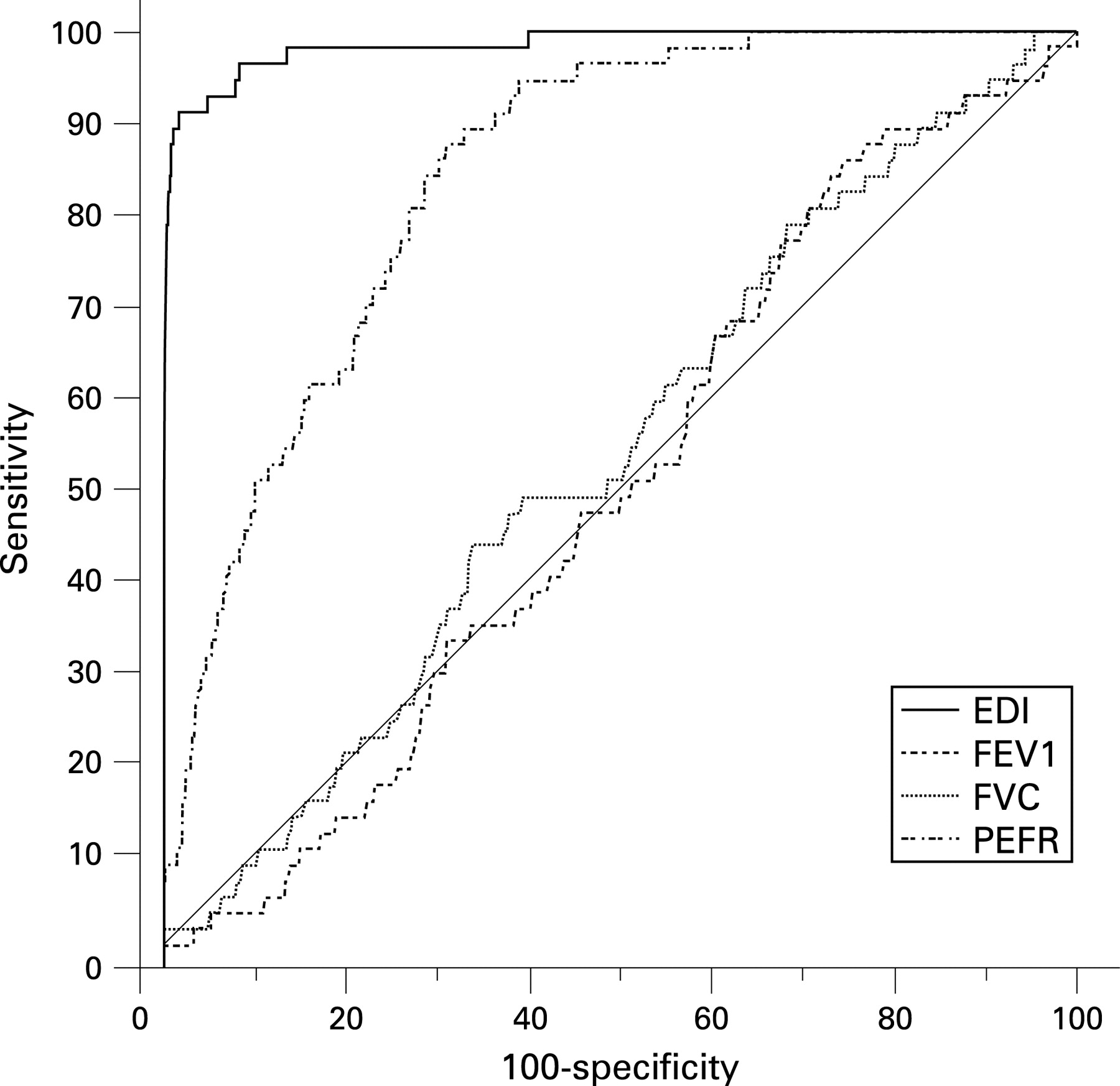
T6 DIAGNOSIS OF LARYNGOTRACHEAL STENOSIS DURING ROUTINE PULMONARY FUNCTION TESTING USING “EXPIRATORY DISPROPORTION INDEX”
1SAR Nouraei, 1K Murphy, 2SM Nouraei, 1DJ Howard, 1GS Sandhu, 3A Patel, 1AR Cummin. 1Imperial College Healthcare NHS Trust, London, UK, 2University of Mazandran, Sari, Iran, 3Royal National Throat Nose and Ear Hospital, London, UK
{kind=link}
Background: Laryngotracheal stenosis is an underrecognised cause of dyspnoea and “wheeze”. Early treatment improves outcome and reduces the need for cervicomediastinal surgery. Conversely, diagnostic failure leads to prolonged treatment for a presumed “resistant” bronchopulmonary diagnosis, which causes significant morbidity and places the patient at ongoing risk of acute-on-chronic potentially fatal airway obstruction. Laryngotracheal stenosis is an uncommon cause of a very common clinical presentation, which creates the challenge of identifying index cases without subjecting many patients to unnecessary and often invasive investigations. To this end we investigated the utility of the expiratory disproportion index (EDI), calculated from routine spirometry variables, in differentiating between laryngotracheal stenosis and other respiratory conditions.
Methods: Diagnostic utility of EDI (FEV1 (ml)/PEFR (l/min)), as described by Empey, was investigated using receiver operator characteristic (ROC) curve and neural networks in 9621 patients, including 3118 volunteers, 3461 patients with chronic obstructive pulmonary disease, 1650 asthmatic patients, 920 patients with pulmonary fibroses, 332 patients with rare lung diseases including sarcoidosis and 140 patients with confirmed laryngotracheal stenosis. The dataset was randomly spliced. One set was used for diagnostic rule extraction and the second for independent validation.
Results: Area under the ROC curve for differentiating laryngotracheal stenosis from other bronchopulmonary diagnoses was 0.985 ± 0.011. At a threshold of 9, sensitivity of 93.4% and specificity of 96.8% were achieved. At a threshold of 8, sensitivity was 96.9% and specificity was 91.1%. Neural networks did not significantly improve detection. There was a significant correlation between residual tracheal lumen cross-section and EDI (r = 0.61; p<0.001) (see fig).
Conclusions: The EDI is a sensitive and specific test that can alone reliably rule in or rule out laryngotracheal stenosis using only routine spirometry. Its clinical utility rests on the principle of disproportionate PEFR diminution relative to FEV1, which almost exclusively occurs with supracarinal resistive loading, as opposed to proportional FEV1–PEF changes that occur in bronchopulmonary diseases. We recommend its use for screening defined at-risk populations such as post-intensive care and Wegener’s granulomatosis patients and suggest more general application in evaluating patients with “unexplained” dyspnoea.