Article Text

Abstract
Background: Neutrophil elastase (NE) activity is increased in lung diseases such as α1-antitrypsin (A1AT) deficiency and pneumonia. It has recently been shown to induce expression of cathepsin B and matrix metalloprotease 2 (MMP-2) in vitro and in a mouse model. It is postulated that increased cathepsin B and MMP-2 in acute and chronic lung diseases result from high levels of extracellular NE and that expression of these proteases could be inhibited by A1AT augmentation therapy.
Methods: Cathepsin and MMP activities were assessed in bronchoalveolar lavage (BAL) fluid from patients with A1AT deficiency, pneumonia and control subjects. Macrophages were exposed to BAL fluid rich in free NE from patients with pneumonia following pretreatment with A1AT. MMP-2, cathepsin B, secretory leucoprotease inhibitor (SLPI) and lactoferrin levels were determined in BAL fluid from A1AT-deficient patients before and after aerosolisation of A1AT.
Results: BAL fluid from both patients with pneumonia and those with A1AT deficiency containing free NE had increased cathepsin B and MMP-2 activities compared with BAL fluid from healthy volunteers. The addition of A1AT to BAL fluid from patients with pneumonia greatly reduced NE-induced cathepsin B and MMP-2 expression in macrophages in vitro. A1AT augmentation therapy to A1AT-deficient individuals also reduced cathepsin B and MMP-2 activity in BAL fluid in vivo. Furthermore, A1AT-deficient patients had higher levels of SLPI and lactoferrin after A1AT augmentation therapy.
Conclusion: These findings suggest a novel role for A1AT inhibition of NE-induced upregulation of MMP and cathepsin expression both in vitro and in vivo.
Statistics from Altmetric.com
Raised levels of proteases are typically observed in acute and chronic lung diseases at sites of inflammation and infection. Proteases have a number of effects including tissue destruction, tissue remodelling and cleavage of soluble innate factors. We have previously shown that cathepsin B can cleave and inactivate key innate immunity proteins including lactoferrin1 and secretory leucoprotease inhibitor (SLPI).2 We have also recently described a novel hierarchy in protease regulation which showed that neutrophil elastase (NE) can induce the expression of cathepsin B and matrix metalloprotease 2 (MMP-2) in vitro and in a mouse model.3 MMPs and cathepsins have previously been found, together with NE, in conditions such as emphysema and cystic fibrosis.4 ,5 We postulated that increased extracellular NE activity in vivo may induce expression of both cathepsins and MMPs. These proteases are capable of significant tissue destruction and inflammation, both directly and indirectly by cleaving key innate anti-inflammatory and antimicrobial proteins. We hypothesised that NE-mediated upregulation of cathepsins and MMPs could be inhibited by α1-antitrypsin (A1AT), a naturally occurring inhibitor of NE. Reduced expression of these destructive proteases could have significant implications in reducing both lung inflammation and cleavage of innate immunity proteins.
In this study we demonstrate that NE upregulates cathepsin B and MMP-2 in vitro and in vivo in pneumonia and A1AT deficiency, and that this effect of NE can be prevented by A1AT. We show increased levels of the key innate immunity proteins lactoferrin and SLPI following A1AT aerosolised augmentation. Protease inhibitors are attractive therapeutic targets in many lung diseases. Inhibition of NE may provide a therapeutic target in conditions with increased cathepsin B and MMP-2 activities such A1AT deficiency, cystic fibrosis, chronic obstructive pulmonary disease and pneumonia. By inhibiting NE, it may be possible to abrogate not just the deleterious effects of this key serine protease, but also the effects of its cysteinyl and metalloprotease secretagogues—cathepsin B and MMP-2, respectively.
METHODS
Selection and description of patients with A1AT deficiency and pneumonia
An augmentation study of aerosolised A1AT in individuals with A1AT deficiency was carried out in the Department of Medicine, University of Florida, Gainesville using plasma purified A1AT produced by Aventis Behring (now ZLB Behring, King of Prussia, PA, USA). A1AT was converted to dry powder by spray drying and packaged in unit dose blisters prior to delivery by a pulmonary delivery system (Nektar, formerly Inhaled Therapeutics, San Carlos, CA, USA). Briefly, a dry powder pulmonary device was used that disperses fine dry respirable powders in a reproducible fashion to the lung. The mass median aerodynamic particle size was 2 μm. This was a dose escalation study evaluating different doses of A1AT from 6 mg to 96 mg given once daily for 14 days.
After adequate sedation and analgesia, bronchoscopy and bronchoalveolar lavage (BAL) were performed with the subject in the supine position. The bronchoscope was wedged into the anterior segment of the right or left upper lobe. BAL was performed in each lobe with five sequential 20 ml aliquots of normal saline solution which were infused quickly with no dwell time between infusion and aspiration. BAL fluid from both lobes was combined and the percentage of the recovered volume was measured. Mucus was removed from the BAL fluid using two layers of sterilised cotton gauze. Cells were separated from the BAL fluid at 2000 rev/min for 15 min. The BAL fluid was then separated and stored at −70°C. The dosage was given once a day for 14 days. Patients were lavaged before entering the study and at a time point after the last aerosol. The pre-BAL day was day −13 or day −14 (ie, 13–14 days before the study) and the post-BAL day was 24 (4) h after the last dose (ie, day 15). For the purpose of this study we evaluated only those individuals with free NE in their BAL fluid before aerosolised augmentation therapy and those for whom we had access to post-aerosolised augmentation BAL samples (6 mg A1AT, n = 11). There was no selection bias before the data were analysed. BAL fluid was obtained according to standardised guidelines and as approved by the Beaumont Hospital and University of Florida Review Board committee. All subjects had a diagnosis of A1AT deficiency, PiZZ phenotype confirmed by nephelometry and isoelectric focusing.
BAL fluid was also obtained from nine patients (five men) with community acquired pneumonia of mean (SD) age 54.0 (1.6) years, of whom three were non-smokers, one was a former smoker for 5–10 years (with a history of 10–20 pack-years) and five were current smokers (10–35 cigarettes/day with a history of 15–30 pack-years). Pneumonia was diagnosed using established criteria. All BAL fluid samples were found to be negative by quantitative culture. Sputum and blood Gram stain and culture were performed. Streptococcus pneumoniae was identified in just under half of the patients. Cytospin preparations of BAL cells were prepared for cytological analysis. Filtered BAL fluid was aliquoted and stored at −80°C. Controls were recruited from medical outpatient clinics at Beaumont Hospital and had no history of pneumonia, current infection, were PiMM phenotype confirmed by nephelometry and isoelectric focusing and were non-smokers (n = 9). Epithelial lining fluid (ELF) levels were determined by measuring urea levels in BAL fluid and serum (from blood taken at the time of bronchoscopy). Ethical approval was obtained and all subjects consented to participate in the study.
Culture and stimulation of monocyte cells
Myelomonocytic cells (U937) (European Collection of Cell Cultures Health Protection Agency, Salisbury, Wiltshire, UK) were cultured in RPMI 1640 medium (Gibco, Paisley, UK) and differentiated to macrophage-like cells for 48 h with phorbol myristic acetate. The macrophage-like cells were incubated in fresh medium for a further 2 days before stimulation. Stimulation was performed with BAL fluid pooled from nine patient samples with pneumonia (mean (SD) NE activity 146.85 (35.06) nM/ELF) for 1 h in fresh serum-free medium, then washed and cultured in fresh serum-free medium for either 3 h or 24 h before harvesting if needed for RNA or protein isolation, respectively.
NE measurements
NE activity in BAL fluid or cell supernatant was determined using the NE-specific substrate N-methoxy-succinyl-Ala-Ala-Pro-Val p-nitroanilide (Sigma, Dublin, Ireland). Release of p-nitroaniline was measured at 405 nm every 2 min over a 20 min time period and was compared with an NE standard of known activity (low-endotoxin elastase derived from human sputum (approximately 50% active); Elastin Products, Owensville, MO, USA).6 All assays were performed in triplicate.
Semiquantitative RT-PCR
After treatment, cells were harvested in Tri reagent (Sigma-Ireland) and RNA was extracted as detailed in the manufacturer’s protocol. RNA (2 μg) was reverse-transcribed at 37°C with 1 mM deoxynucleotide mix (Promega, Southampton, UK), 1.6 μg oligo-p[dT]15 primer (Roche, Lewes, UK) and 1 μl M-MLV reverse transcriptase (Promega) in a 20 μl volume as described in the manufacturer’s protocol. 2 μl of each cDNA was amplified with 1.25 U Taq DNA polymerase, 1×PCR buffer and 10 mM dNTPs (Promega) in a 50 μl volume containing 100 pmol each of the following primers: 5′-ATG TGG CAG CTC TGG GCC T-3′ and 5′-TAC TGA TCG GTG CGT GGA ATT-3′ for cathepsin B; 5′-GCC CCC AAA ACG GAC AAA GA-3′ and 5′-TCC CAA GGT CCA TAG CTC ATC G-3′ for MMP-2; 5′-AAC TCT GGT AAA GTG GAT-3′ and 5′-TAC TCA GCG CCA CCA GCA TCG-3′ for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). PCR products were quantified densitometrically at cycle numbers between 10 and 40 to determine the appropriate cycle number at which exponential amplification of products was occurring, and to identify the cycle number at which sufficient discrimination was possible to accurately quantify increases or decreases in gene expression. After a hot start the amplification profile was 32 cycles of 1 min denaturation at 94°C, 1 min annealing at 58°C and 1 min extension at 72°C. RT-PCR amplification of cathepsin B, MMP-2 and GAPDH generated products of 1004 bp, 525 bp and 211 bp, respectively. PCR products were commercially sequenced (MWG Biotech AG, Ebersberg, Germany) to verify gene identity. PCR products were resolved on a 1% agarose gel containing 0.5 μg/ml ethidium bromide (Sigma). The ratio of PCR fragment intensities of cathepsin B and MMP-2 relative to GAPDH was determined by densitometry.
Presence of cathepsin B
Cathepsin B activity was determined from either BAL fluid or medium taken from macrophage-like cells 24 h after stimulation with or without BAL in 100 μl of each sample using the substrate Z-Arg-Arg-AMC (0.1 mM). A cathepsin B inhibitor CA-074 (10 μg/ml) was used as a control for the specificity of the cathepsin B substrate. The reaction buffer used for cathepsin B activity estimation was 0.2 M sodium acetate, 2 mM EDTA, 1 mM DTT, 1 μM pepstatin and 2 mM Pefabloc, pH 5.5. The samples were incubated with substrate for 60 min at 37°C and fluorescence (substrate turnover) was determined by excitation at 355 nm and emission at 460 nm. The results were expressed as a change (delta) in fluorescence units (FU) over a 60 min period.
Zymography
Gelatin zymography was performed on either BAL fluid samples or medium collected from unstimulated or BAL fluid-stimulated cells. Samples were subjected to 7% SDS-polyacrylamide gel electrophoresis with a gel-containing gelatin (1 mg/ml). After electrophoresis, the gels were incubated in 50 mM Tris (pH 7.5), 5 mM CaCl2, 1 μM ZnCl and 2.5% (v/v) Triton X-100 for 30 min, washed in the same buffer without the Triton X-100 for 5 min, then incubated at 37°C overnight in the same buffer supplemented with 1% (v/v) Triton X-100. The gels were stained with 0.125% Coomassie Blue and washed with 10% acetic acid and 40% methanol in water. The presence of MMPs appears as transparent bands. Latent MMP-2 and active MMP-2 were observed at 72 and 66 kDa, respectively. Densitometry was carried out to compare the intensity of the MMP transparent bands.
Western blot analysis
BAL fluid from healthy controls and A1AT-deficient patients was investigated for the presence of cathepsin B. BAL fluid samples were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane (Sigma-Aldrich, Dublin, Ireland) which was probed using rabbit anti-cathepsin B antibody (Calbiochem, Nottingham, UK). Binding was detected using the appropriate horseradish peroxidase-conjugated secondary antibody and visualised by chemiluminescence (Pierce, Dublin, Ireland).
Densitometric analysis
Gels were analysed by densitometry and compared in a semiquantitative manner using the GeneGenius Gel Documentation and Analysis System (Cambridge, UK) and GeneSnap and GeneTools software. All expression values were verified by at least two independent experiments.
Statistical analysis
Data were analysed with the PRISM 3.0 software package (GraphPad, San Diego, CA, USA). The results are expressed as mean (SE) and were compared by t test. When more than two groups were being compared, an ANOVA test was used followed by a Tukey post hoc test. Differences were considered significant at p values ⩽0.05.
RESULTS
BAL fluid characteristics
BAL fluid samples were collected from patients with A1AT deficiency, pneumonia and control subjects. Cytospin preparations were analysed to determine cell numbers/ml BAL fluid. The sample data for the A1AT-deficient patients are summarised in table 1. NE activity levels were reduced from 297.0 (325.4) to 0.00 nM/ELF following A1AT aerosolised augmentation therapy, but without reducing the percentage of neutrophils in the BAL fluid. Samples from pneumonia-infected lungs contained increased numbers of cells compared with controls (1.59×106 (8.3×105) cells/ml vs 3.89×105 (7.6×104) cells/ml, p<0.05). The macrophage:neutrophil ratio in the infected ELF was 5:3 compared with 4:1 in uninfected ELF. Pneumonia was characterised by a neutrophil influx with 5.5-fold more neutrophils at the infection site for control and infected subjects (8.4×104 (6.2×103) cells/ml and 4.7×105 (2.4×104) cells/ml, p<0.05).
Protease profile of BAL fluid
Cathepsin B and MMP-2 activities were measured in BAL fluid from individuals with A1AT deficiency and those with pneumonia. Increased cathepsin B activity was observed in BAL fluid from individuals with A1AT deficiency compared with BAL fluid from non-smoking healthy controls using the cathepsin substrate Z-Arg-Arg-AMC (fig 1A). Incubation of BAL fluid from A1AT-deficient subjects with the cathepsin B inhibitor CA-074 significantly reduced turnover of the substrate Z-Arg-Arg-AMC, indicating that cathepsin B is the primary cathepsin present in BAL fluid from individuals with A1AT deficiency (fig 1A, p<0.001 when all experimental groups were compared). This was further confirmed by Western blot analysis which demonstrated the presence of cathepsin B in BAL fluid from subjects with A1AT deficiency but not in BAL fluid from healthy control subjects (fig 1B).

Cathepsin B activity was higher in BAL fluid from patients with pneumonia than in BAL fluid from controls (fig 1A, p<0.001). Active forms of MMP-2 were present in BAL fluid from patients with A1AT deficiency and pneumonia but not in BAL fluid from control subjects (fig 1C).
Cathepsin B and MMP-2 gene expression and activity in macrophages exposed to BAL fluid from individuals with pneumonia containing free NE
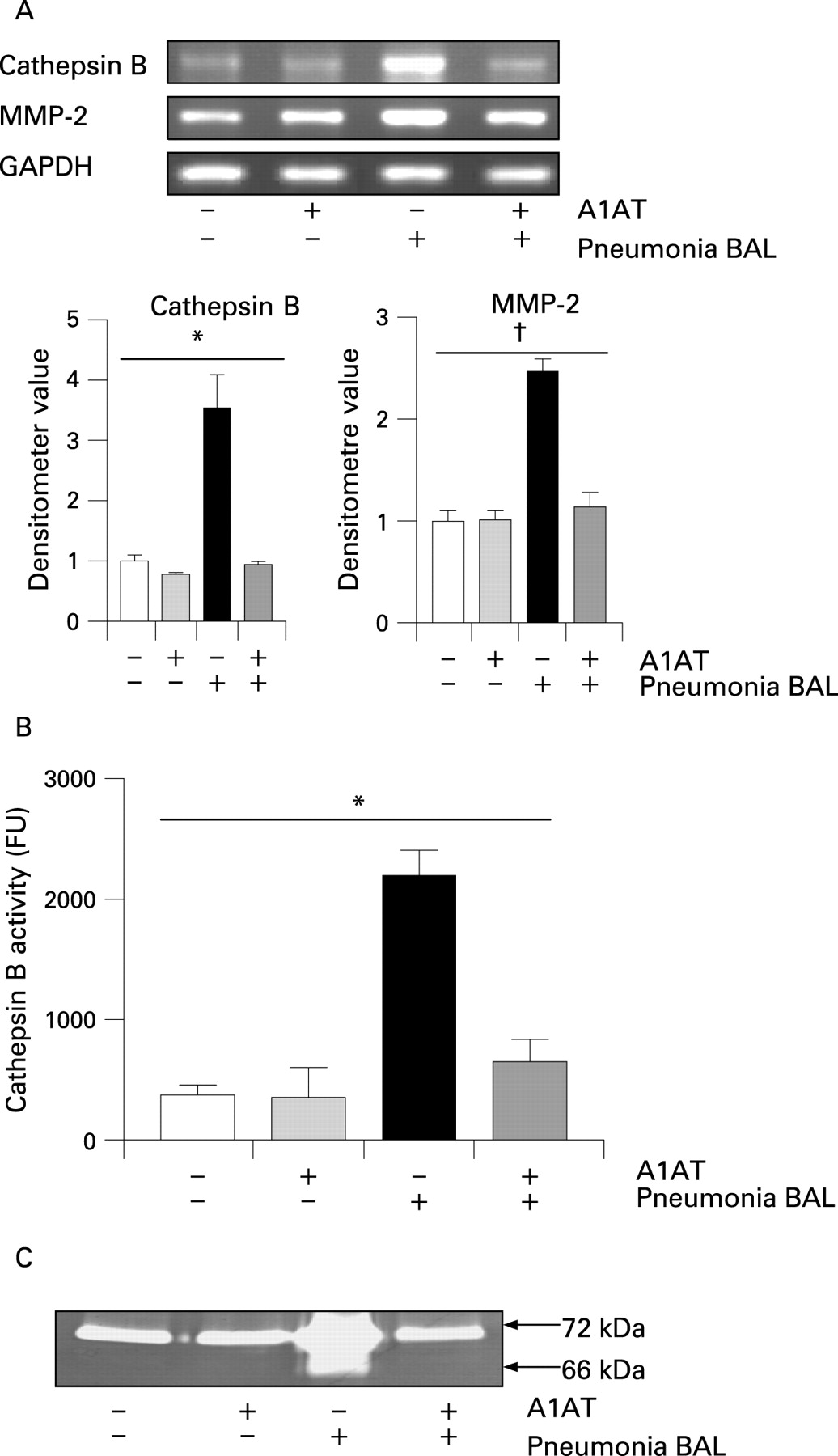
Previous investigators have used U937 cells to investigate MMP and cathepsin activity extracellularly secreted from monocytes and macrophages.7 ,8 To examine whether exposure of differentiated U937 cells to BAL fluid samples obtained from patients with pneumonia could mimic the effect of NE, differentiated U937 cells were exposed to BAL fluid from individuals with pneumonia containing active NE (146.85 (35.06) nM). Cathepsin B and MMP-2 mRNA expression levels were investigated by RT-PCR (fig 2A). Cathepsin B (p = 0.006) and MMP-2 (p = 0.002) gene expression increased when cells were stimulated with BAL fluid from patients with pneumonia, but this increased expression was suppressed when cells were pretreated with A1AT. No NE activity was recorded in BAL fluid from individuals with pneumonia or cell supernatant following addition of A1AT (data not shown). Cathepsin B and MMP-2 activities were measured in the supernatants, and stimulation with BAL fluid from individuals with pneumonia was found to increase cathepsin B activity compared with non-stimulated control cells (fig 2B). Stimulation with BAL fluid from individuals with pneumonia resulting in cathepsin B activity was inhibited when cells were pretreated with A1AT (p<0.001 when all experimental groups were compared). Increased active MMP-2 was observed when cells were treated with BAL fluid from individuals with pneumonia (fig 2C), which was abrogated when the BAL fluid from these individuals was incubated with A1AT.

Protease and antiprotease activity in patients with A1AT deficiency following aerosolised augmentation therapy
Cathepsin B and MMP-2 activity was measured in BAL fluid from 11 patients with A1AT deficiency before and after A1AT treatment. Patients were treated with 6 mg A1AT and BAL fluid was recovered 12 h later. Cathepsin B activity was significantly decreased when patients were treated with A1AT (fig 3A, p = 0.002). A1AT treatment also resulted in a statistically significant reduction in latent and active MMP-2 (fig 3B, p = 0.039 and 0.040, respectively). The band observed at 78 kDa is active MMP-9.

{kind=link}
{kind=link}
{kind=link}
SLPI and lactoferrin levels in BAL fluid from patients with A1AT deficiency before and after A1AT treatment were examined by ELISA (fig 3C). Lactoferrin levels were significantly increased following A1AT treatment (p = 0.035), while SLPI levels were increased but not significantly (p = 0.286).
DISCUSSION
The pulmonary extracellular matrix is a constantly changing infrastructure comprised of a complex mixture of proteoglycans, collagen and elastin. It is estimated that as much as one-tenth of the extracellular matrix is degraded and replaced each day in the normal lung.9 Members of the serine, matrix metalloprotease and cysteinyl protease families have been associated traditionally with lung inflammation and airway extracellular matrix destruction. Cathepsin B is a cysteinyl protease that is expressed ubiquitously in lung tissue, particularly in bronchial epithelial cells and macrophages.10 Cathepsin B degrades types IV and X collagen and also fibronectin in vitro,11 ,12 and intratracheal instillation of cathepsin B has been shown to induce emphysema in hamsters.13 Cathepsin B activity is also increased in infective and inflammatory conditions such as pneumonia and cystic fibrosis. Schmidt et al14 found increased cathepsin B activity in BAL fluid from patients with both chronic bronchitis and pneumonia. We have recently shown that cathepsin B activity in BAL fluid from patients with cystic fibrosis is increased several hundred fold compared with BAL fluid from healthy subjects.5
A number of secretagogues of cathepsin B have been described. For example, interferon γ causes emphysema and alterations in the pulmonary protease/antiprotease balance when expressed in pulmonary tissues in mice by inducing cathepsins B, H, D, S and L as well as several MMPs. Furthermore, this inflammation can be attenuated by cathepsin inhibition.15 Cigarette smoke has been shown to stimulate cathepsin B activity in alveolar macrophages of rats.16 Lipopolysaccharide has been shown to accelerate caspase-independent but cathep-sin B-dependent cell death of human lung epithelial cells, and inhibition of cathepsin B was shown to prevent lung cell apoptosis. Recently, cathepsin B was one of several cathepsins implicated in the pathogenesis of bronchopulmonary dysplasia.17 Clearly, cathepsin B is a key protease involved in a number of lung diseases. Work by Burnett et al18 showed that the predominant form of cathepsin B (Mr 42 000) is converted to an active form (Mr 38 000) on treatment with NE, and we have shown that extracellular NE is not only required to activate cathepsin B.3 The results of the current study demonstrate similar findings in BAL fluid from human subjects, and suggest a novel way potentially to prevent cathepsin B- and MMP-2-mediated lung inflammation and destruction by inhibiting their upregulation by NE.
Similarly to cathepsins, overexpression of MMPs has been associated with destruction in the lung.19 MMP-2 has been found in pneumocytes, fibroblasts and alveolar macrophages in both emphysematous and normal lung tissue samples, with significantly more immunoreactivity in the emphysematous samples.20 Interleukin 13 (IL-13) upregulates MMP-2 and several other MMPs as well as cathepsins to cause emphysema in a mouse model.15 ,21
We have recently shown that NE can induce cathepsin B and MMP-2 expression in macrophages and a mouse model of Pseudomonas infection.3 We demonstrated that NE can activate NF-κB in macrophages, and inhibition of NF-κB or TLR-4 or transfection of macrophages with dominant-negative IL-1R-associated kinase-1 resulted in a reduction in NE-induced cathepsin B and MMP-2. Given that these proteases are responsible for much of the lung inflammation and destruction in acute and chronic lung diseases, we postulated that inhibition of NE might abrogate their upregulation and production. Inhibition of the ability of NE to upregulate such key destructive proteases could provide potential new strategies for therapeutic interventions.
A1AT is the principal inhibitor of NE. Deficiency of A1AT in the lung results in insufficient anti-elastase protection in the lower respiratory tract, thus allowing NE to destroy alveolar structures leading to premature and aggressive emphysema. Inhibition of NE in the A1AT-deficient lung has been investigated as a therapeutic target for the past 20 years. The goal of A1AT aerosolised augmentation therapy in A1AT deficiency is to raise lung A1AT levels and anti-NE capacity, thereby providing adequate protection against NE and preventing elastase-mediated degradation.22 Previous augmentation studies have used intravenous and aerosol delivery systems. Both modes of delivery have shown increased A1AT levels and anti-NE capacity in the lung ELF. Similar to the inflammatory manifestations of A1AT deficiency, neutrophil-dominated inflammation on the respiratory epithelial surface also occurs in cystic fibrosis resulting in a chronic epithelial burden of NE. A1AT has been given by aerosol form to patients with cystic fibrosis and has successfully suppressed NE in ELF and restored the ELF anti-NE capacity.23 This treatment also reversed the inhibitory effect of cystic fibrosis ELF on Pseudomonas killing by neutrophils, which suggested that it may augment host defence in cystic fibrosis.
Replacement therapy for A1AT deficiency-induced emphysema may be associated with a reduction in the frequency and severity of lung infections as well a reduction in inflammation.24 ,25 Lactoferrin and SLPI have key anti-inflammatory and antimicrobial roles. Lactoferrin has been shown to inhibit the lipopolysaccharide-induced expression and proteoglycan binding ability of IL-8 in human endothelial cells.26 Lactoferrin can bind unmethylated CpG motifs, which are bacterial DNA products capable of stimulating various innate and acquired immune responses in human and murine models.27 It is bactericidal against many of the common pathogens seen in the lung, is fungicidal and can prevent viral replication.28–30 SLPI possesses many similar attributes, having important antibacterial, antiviral and anti-inflammatory properties. SLPI can inhibit lipopolysaccharide-induced nuclear factor-κB activation by inhibiting degradation of interleukin 1-receptor associated kinase, inhibitory (I) KBκ and IκBβ, and can also impair lipotichoeic acid- and lipopolysaccharide-induced proinflammatory gene expression in monocytes and macrophages in vitro.31 ,32 Lactoferrin and SLPI have been shown to act synergistically in bacterial killing.33
In this study, augmentation with A1AT resulted in increased levels of lactoferrin in BAL fluid from patients with A1AT deficiency. We speculate that aerosolised augmentation therapy resulted in decreased cathepsin cleavage of these key anti-inflammatory and antimicrobial proteins. Restoration of host innate immunity by abrogating cathepsin cleavage suggests a further potential therapeutic effect of aerosolised augmentation therapy.
The activities of multiple proteases in chronic lung disease are clearly deleterious to the host. We have elucidated a hierarchy in protease control and regulation whereby NE orchestrates tissue destruction and inflammation by activation of other proteases (cathepsin B and MMP-2.) Neutralisation of the effects of NE with a specific antiprotease (A1AT) may be sufficient to lessen the overall protease burden in chronic infective lung disease without the need for inhibition of all proteases. This may not only prevent protease-induced lung destruction, but may play an important role in restoring lung host defence.
REFERENCES
Footnotes
Funding: This work was supported by the Alpha One Foundation, Health Research Board of Ireland, the Program for Research in Third levels Institutes administered by HEA, Science Foundation Ireland, Cystic Fibrosis Hope Source, Cystic Fibrosis Research Trust, Cystic Fibrosis Association of Ireland and the Royal College of Surgeons in Ireland.
Competing interests: None.
Linked Articles
- Airwaves