Article Text

Abstract
Background: Mutations in the ABCA3 gene can result in fatal surfactant deficiency in term newborn infants and chronic interstitial lung disease in older children. Previous studies on ABCA3 mutations have focused primarily on the genetic abnormalities and reported limited clinical information about the resultant disease. A study was undertaken to analyse systematically the clinical presentation, pulmonary function, diagnostic imaging, pathological features and outcomes of children with ABCA3 mutations.
Methods: The records of nine children with ABCA3 mutations evaluated at Texas Children’s Hospital between 1992 and 2005 were reviewed and their current clinical status updated. Previous diagnostic imaging studies and lung biopsy specimens were re-examined. The results of DNA analyses were confirmed.
Results: Age at symptom onset ranged from birth to 4 years. Cough, crackles, failure to thrive and clubbing were frequent findings. Mean lung function was low but tended to remain static. CT scans commonly revealed ground-glass opacification, septal thickening, parenchymal cysts and pectus excavatum. Histopathological patterns included pulmonary alveolar proteinosis, desquamative interstitial pneumonitis and non-specific interstitial pneumonitis, and varied with age. Dense abnormalities of lamellar bodies, characteristic of ABCA3 mutations, were seen by electron microscopy in all adequate specimens. Outcomes varied with the age at which the severity of lung disease warranted open lung biopsy, and some patients have had prolonged survival without lung transplantation.
Conclusions: The presentation and course of interstitial lung disease due to ABCA3 mutations are variable, and open lung biopsy and genetic testing are warranted early in the evaluation of children with a consistent clinical picture.
Statistics from Altmetric.com
An important development in the field of childhood interstitial lung disease (chILD) has been the discovery of inborn errors of surfactant metabolism, including mutations in the surfactant protein B (SP-B), surfactant protein C (SP-C) and ABCA3 genes.1–3 Mutations in these genes cause chILD with varying histopathological patterns including desquamative interstitial pneumonitis (DIP), chronic pneumonitis of infancy, pulmonary alveolar proteinosis (PAP) and non-specific interstitial pneumonitis (NSIP).1–10 ABCA3 is an ATP-binding cassette transporter of lipids that is found in the limiting membrane of lamellar bodies in alveolar type II cells.11 Initially reported as a cause of fatal lung disease in term neonates with a clinical picture similar to that of SP-B deficiency,3 mutations in the ABCA3 gene are now known also to cause chronic interstitial lung disease (ILD) in older patients.8 Previous reports on ABCA3 mutations focused primarily on the genetic abnormalities and gave limited clinical information about the resultant disease beyond the neonatal period. We present an analysis of nine children with ABCA3 mutations evaluated at a single medical centre in order to describe more fully the clinical presentations, pulmonary function, diagnostic imaging, pathological features and outcomes of children with this disorder.
METHODS
Nine children who were eventually diagnosed with mutations in the ABCA3 gene were evaluated at Texas Children’s Hospital (TCH), a tertiary chILD and pediatric lung transplantation referral centre, between 1992 and 2005. Five of the patients were transferred to or primarily cared for at our centre while four were seen on an outpatient referral basis. The records of patients were reviewed to document the neonatal history, interval history prior to initial evaluation at TCH, presenting symptoms, family and environmental history, physical findings at initial evaluation, pulmonary function tests and treatment regimen. Each patient’s current clinical status—including growth parameters, pulmonary function and ILD score12—was obtained from clinical records and communications with parents and referring physicians. Records of the initial evaluation were available and adequate for analysis in each case, and the clinical status was updated for all survivors. All available chest radiographs, CT scans and lung pathology specimens were systematically re-examined (without blinding) by a paediatric radiologist (RPG) and a paediatric lung pathologist (MKD), respectively, at TCH. DNA samples from seven patients were initially analysed for ABCA3 mutations by direct sequence analysis of the 30 coding exons as previously described under a research protocol at the Johns Hopkins University School of Medicine,8 and their mutations were subsequently confirmed in the CLIA certified Johns Hopkins DNA Diagnostic Laboratory where the mutations of the remaining two patients were also identified. Two patients (1 and 6) have been described in previous publications.8 13
RESULTS
Clinical presentation
All nine patients in our cohort were born at term. Five had respiratory symptoms in the newborn period, including three patients who developed respiratory failure (two were extubated after 2 days; one had progressive deterioration) and two patients who were treated for pneumonia with intravenous antibiotics and/or oxygen. In the four patients who were eventually discharged home, three continued to have either persistent or intermittent respiratory symptoms (described below), while one infant was apparently well until 8 months of age. In the other four children who had an unremarkable neonatal course, the age at symptom onset ranged from 3 months to 4 years. Known environmental exposures were present in three patients: one to birds and two to cigarette smoke. A family history of chILD was absent at the time of initial evaluation in all nine cases.
The eight patients who did not have persistent respiratory failure from the newborn period all presented later with cough, tachypnoea, dyspnoea and exercise intolerance (table 1). Most had hypoxaemia at rest, while one had hypoxaemia only with exertion. On initial evaluation at our centre, the majority of patients were below the 5th percentile for weight and had crackles and retractions on examination. Only one child had wheezing. The seven children who were older than 2 years at initial evaluation all had clubbing, and five of them also had pectus excavatum apparent on physical examination.
Pulmonary function tests
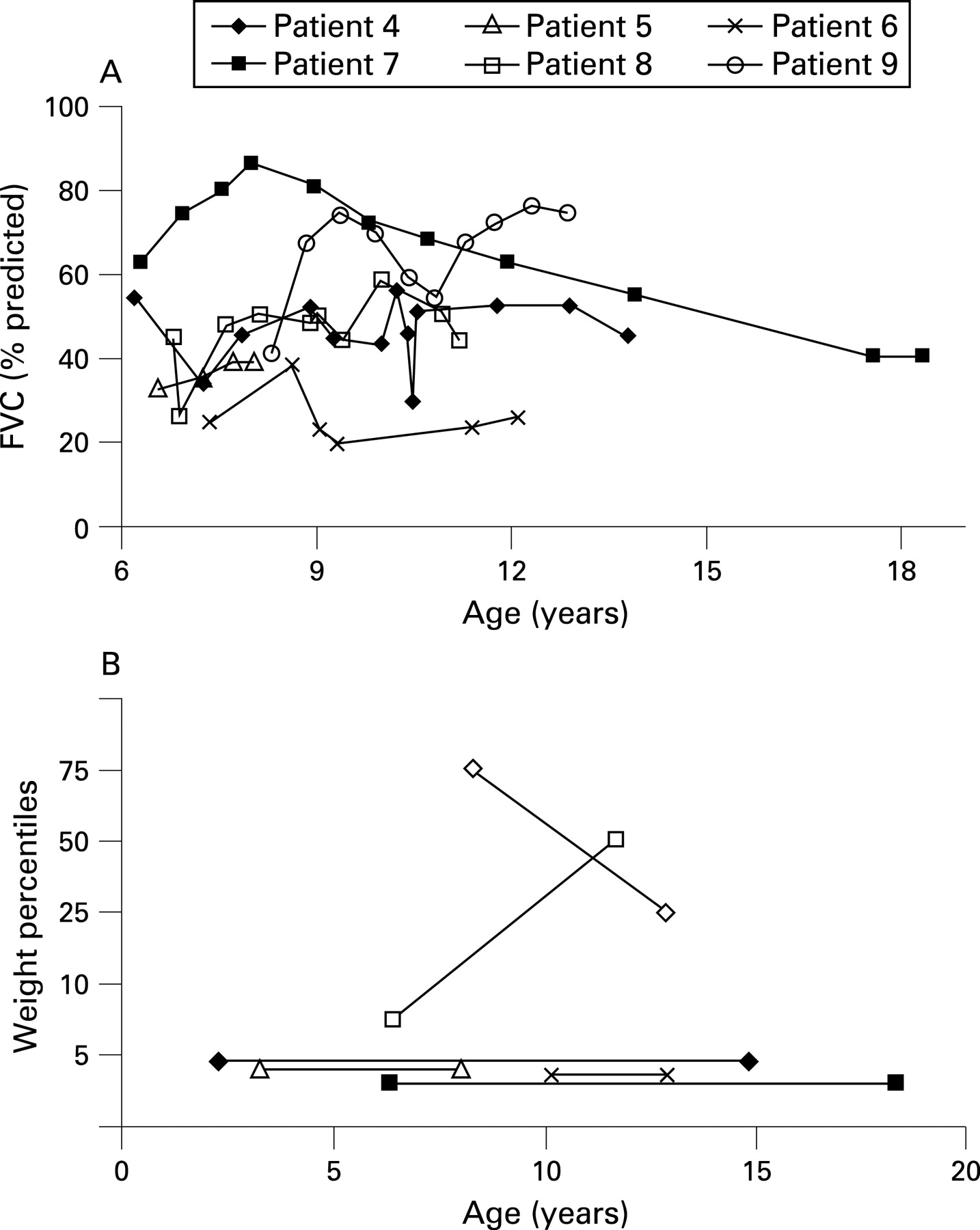
Six patients were able to perform spirometry starting at age 6–8 years (fig 1A). The mean (SD) initial forced vital capacity (FVC) and forced expiratory volume in 1 s (FEV1) percentage predicted were 43.6 (13.9)% and 41.2 (12.0)%, respectively, consistent with severe restrictive lung disease. Most of the patients were unable to perform the manoeuvre for measurement of the carbon monoxide lung transfer factor (Tlco).

Diagnostic imaging
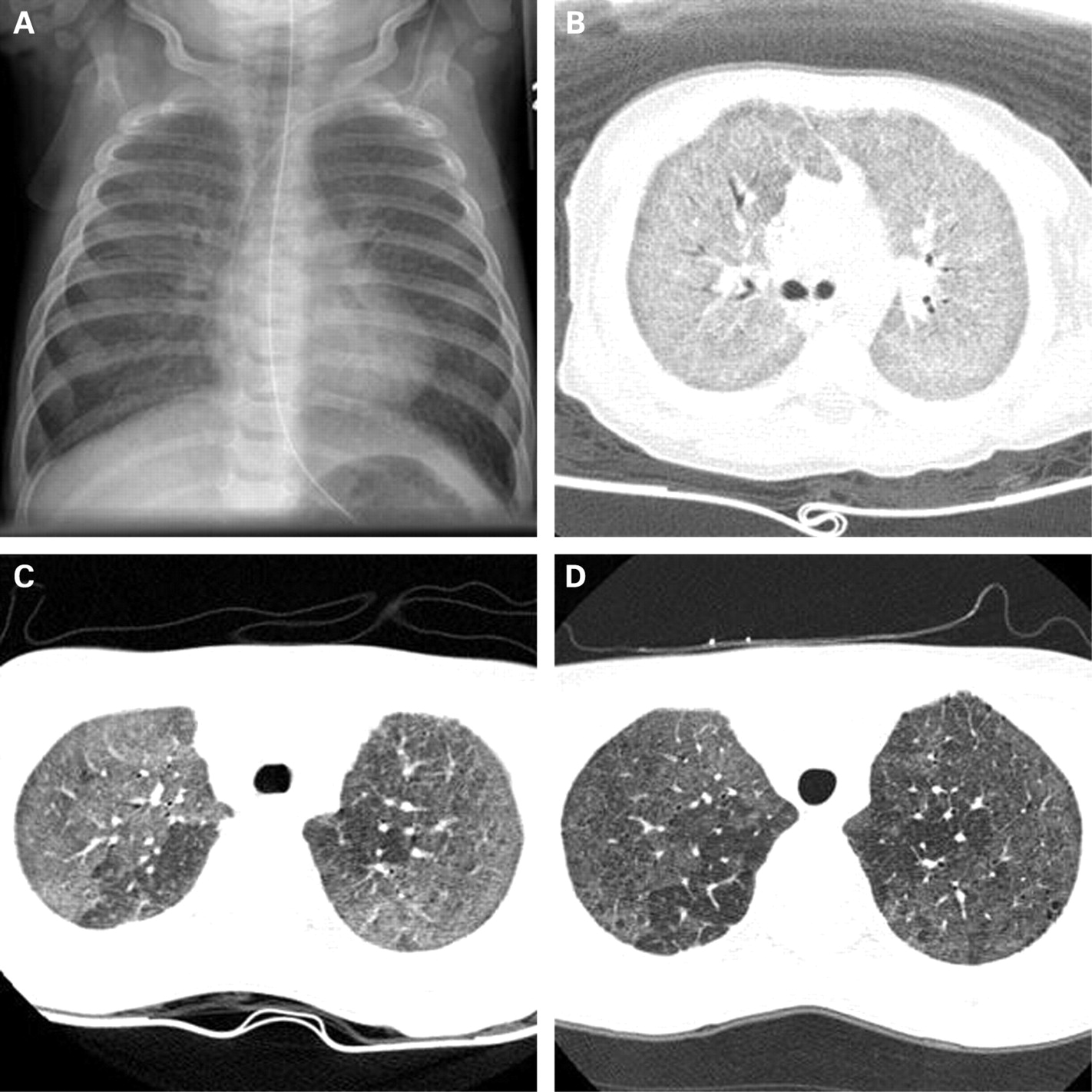
Chest radiographs were available for review for all nine patients at various ages. They consistently showed bilateral diffuse or patchy hazy granular or streaky opacities (fig 2). One or more helical or high-resolution axial CT (HRCT) scans were available for review in eight patients; the ninth was too critically ill to undergo CT imaging. On CT scan, all patients had ground-glass opacification that was either diffuse throughout the lungs or patchy but involving multiple or all lobes. The imaging appearance during infancy is very similar to neonatal respiratory distress syndrome. In some patients the intensity of the ground-glass opacification decreased with age, but this did not correlate with improvement in lung function or degree of hypoxaemia. All patients imaged beyond infancy developed fine or coarse peripheral interstitial septal thickening. Five patients eventually developed small (few mm in diameter) air-filled parenchymal lung cysts that tended to increase in number and size over time. Hilar and mediastinal lymphadenopathy (four patients), consolidation and atelectasis that cleared over time (four patients), pleural thickening (three patients) and air trapping (three patients) were also seen. Pectus excavatum (defined as Haller index >2.7)14 developed in all patients surviving beyond infancy.
Pathology
Diagnostic lung biopsy slides were available for review in all nine patients, including three with a second biopsy and two with subsequent lung pathology specimens after transplantation and at autopsy. The time interval between specimens was <1 month for three patients and similar histological features were seen in the paired specimens in each case. Two subjects (patients 1 and 6) had an extended interval between biopsies (4 years in both cases), and these biopsies are considered separately. Histological features and major patterns of disease for these 11 biopsies are summarised in table 2. The spectrum of overlapping histological findings were grouped into three major categories: PAP pattern, DIP pattern and NSIP pattern (fig 3). Features present in all 11 cases included some degree of lobular remodelling, at least focally increased alveolar macrophages and at least focal pneumocyte hyperplasia. All cases showed either alveolar proteinosis material or cholesterol clefts (endogenous lipoid pneumonia), with both features present in five biopsies. Alveolar proteinosis material and diffuse alveolar epithelial hyperplasia were most prominent in the infants and were inconspicuous in the older children at the time of biopsy. Iron-laden macrophages were present in all patients except for the two youngest (aged 3 weeks and 1 month). Lobular remodelling was most prominent in the older children, although this finding was variable with some biopsies showing a patchy distribution of architectural abnormalities.

The two patients with follow-up biopsies at prolonged intervals allowed for examination of the progression of histological findings over time. When a biopsy specimen was taken at 3 weeks of age, patient 1 initially had a PAP pattern with early lobular remodelling and a reactive widened interstitium (fig 4A and B). At follow-up biopsy 4 years later, the lung showed more extensive lobular remodelling (airspace enlargement and increased interstitial smooth muscle), mildly increased interstitial inflammatory cells, but less reactive interstitial widening (fig 4C and D). Alveolar proteinosis was present in both biopsies, with the additional finding of endogenous lipoid pneumonia at 4 years of age. Patient 6 underwent biopsies at 4 years (fig 4E) and 9 years of age (fig 4F), both showing an NSIP pattern with endogenous lipoid pneumonia and extensive lobular remodelling. Both specimens showed increased lymphoid infiltrates and cholesterol clefts, although these findings were more prominent in the second biopsy.

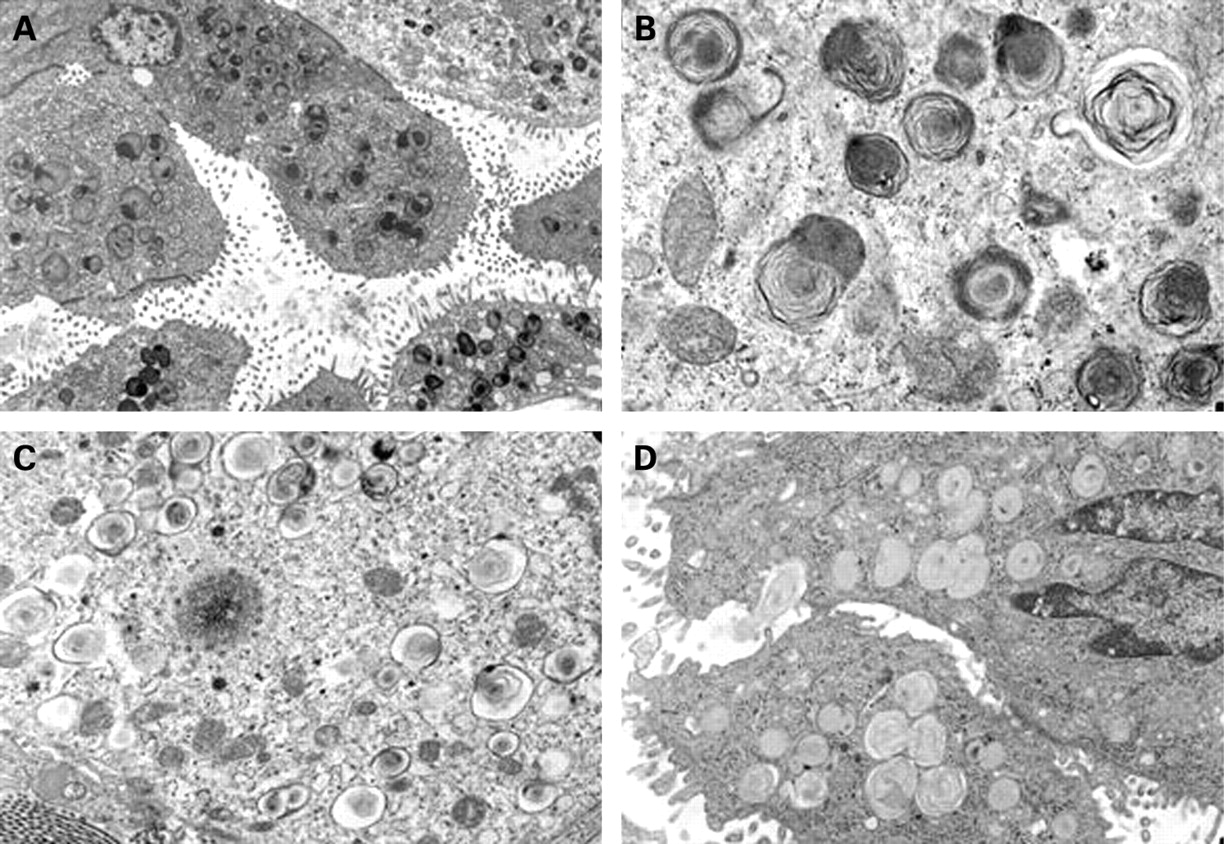
Tissue was available for electron microscopic study in six biopsies, with adequate preservation of the ultrastructure in five cases. Characteristic small lamellar dense bodies were identified in all five patients but were more frequent and well formed in the younger children (age range 3 weeks to 2 years) and inconspicuous in the one biopsy from an older child (age 6 years) (fig 5). The dense bodies varied in size and location, including both centrally positioned and eccentric round electron-dense forms. The lamellar structure associated with the dense bodies was delicate and whorled in some areas and more compact with increased electron density in other areas. In addition to these dense bodies, the type II alveolar epithelial cells also showed increased lysosomal material, forming dense black globular cytoplasmic aggregates focally containing components of lamellar membranes, suggesting intralysosomal degeneration of abortive lamellar bodies. Composite lamellar bodies with apparent fusion of two or three whorled lamellar bodies were also detected at least focally in each case. The abnormal dense bodies and degenerating lysosomal lamellar structures were also admixed with a few lamellar bodies of normal size and structure in each case, and these normal lamellar forms predominated in the oldest child (age 6 years).
Genetics
There were eight Caucasians and one Hispanic in the cohort. DNA analysis for mutations in the ABCA3 gene revealed two mutations in all nine patients consistent with autosomal recessive inheritance. Five of these mutations (R20L, ΔF1203, E292V, T114M, Q1591P) have been previously identified in subjects with chILD, whereas the remainder are novel. The E292V mutation was the only one found in more than one patient. Eight patients had been followed for many years with a diagnosis of idiopathic chILD, while one (patient 1) was prospectively diagnosed at 2 months of age.
Treatment and outcome
All nine patients were treated with systemic corticosteroids, seven with pulses of intravenous methylprednisolone given on a monthly basis for varying durations. Hydroxychloroquine was added to the treatment regimen of seven patients. Records were not sufficiently complete to determine the short-term response to treatment. Long-term outcomes within our cohort were variable. One infant died from unrelenting respiratory failure at 4 months of age, 1 month after initial presentation (table 1). Three patients underwent lung transplantation at ages 3 months, 5 years and 12 years; the youngest was the child with persistent respiratory failure from the neonatal period. The two youngest transplant recipients have died (one from primary graft dysfunction and one from bronchiolitis obliterans), while the oldest is still alive <1 year after transplantation.
The five survivors who have not required transplantation currently range in age from 8 to 18 years. Three of them have had an improvement in the degree of their hypoxaemia (as evaluated by ILD score) while two have not. None have evidence of secondary pulmonary arterial hypertension by electrocardiography or echocardiography. The lung function in four of these five patients has not changed significantly over time; the fifth has had a slow decline from a peak FEV1 of 80% to 40% predicted over 10 years, although he currently reports no symptoms and is very active physically. The mean (SD) latest FVC and FEV1 were 48.9 (14.8)% predicted and 48.1 (18.5)% predicted, respectively, in the five survivors without transplant. In the six patients who are still alive, all four who had a weight <5th percentile at the initial evaluation are still at <3rd percentile for weight, while the two children who did not have failure to thrive have continued to have normal growth with follow-up intervals ranging from 2 to 12 years (fig 1B).
Within our cohort no specific clinical feature correlated with outcome, as measured by the need for lung transplantation or death, although these analyses were difficult because of the small number of patients. There was a suggestion that younger age at the time of initial lung biopsy might indicate a lower likelihood of surviving without transplantation (fig 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
Our study is the first to present a systematic review of the clinical features, lung function, imaging changes and histopathology of children with ABCA3 mutations. The results of our analysis extend the descriptions of previous studies and reveal several clinically significant findings not previously described.
Although ABCA3 mutations are inherited in an autosomal recessive fashion,3 none of our patients had a positive family history at the time of presentation, underscoring the need to consider this entity even in the absence of such history. The higher proportion of cases with a positive family history reported in previous studies may reflect selected patient populations.3 8 10 The age at symptom onset for nearly half of our patients was beyond the neonatal period and extended to early childhood, re-emphasising the findings of Bullard et al8 that ABCA3 mutations can present as chronic ILD in young children.
This study provides the first description of the serial lung function, growth and radiographic changes in children with ABCA3 mutations. The FVC percentage predicted, while initially low, tended to remain stable over many years. Among the survivors without lung transplantation, the majority had a mild improvement in oxygenation. Most of the children who presented with chronic ILD beyond the neonatal period had failure to thrive, and all of these patients continued to grow poorly. The remainder, who underwent open lung biopsy at older ages (suggesting milder disease), did not have growth abnormalities at initial evaluation and they continued to grow well. Serial CT imaging of the lungs consistently showed bilateral ground-glass opacification that in some cases decreased in intensity with age, while peripheral interstitial thickening and small parenchymal lung cysts became more prominent findings over time. Interestingly, pectus excavatum was seen in all patients who were imaged beyond infancy; one child underwent surgical correction of his pectus before he was diagnosed with ABCA3 mutations. We are aware of only one previous report of pectus excavatum in association with chronic ILD in a patient with pulmonary alveolar microlithiasis.15 Changes in CT findings over time did not correlate with lung function, hypoxaemia or outcome, suggesting that once the diagnosis is established, frequent routine imaging studies are not warranted.
The systematic review of all open lung biopsy specimens in our series—including repeat biopsies at older ages in two children—showed that the pathological phenotypes of ABCA3 mutations are age-dependent. In the biopsy specimens obtained from young infants, PAP and DIP were the predominant patterns. In the two biopsy specimens from toddlers, one showed a DIP pattern and the other had an NSIP pattern. Finally, the older children—who underwent biopsy at 4–9 years of age—all had an NSIP pattern, often with superimposed endogenous lipoid pneumonia. No cases showed a complete constellation of features seen in chronic pneumonitis of infancy. As this pattern is most often recognised in older infants (6–18 months) with SP-C mutations, its absence in our series may reflect this gap in age distribution. While different histological patterns have been reported in previous studies of ABCA3 mutations,3 8–10 an age-dependent variation in such patterns has not been described. Together with earlier reports, our findings emphasise that disease-causing mutations in the ABCA3 gene can lead to multiple pathological diagnoses. It is important to note that several of our patients had been followed for many years (in one case into adulthood) with the pathological diagnosis of NSIP or DIP, raising the possibility that some adults with long-standing chronic ILD who have these pathological patterns may actually have ABCA3 mutations.
The presence of characteristic small lamellar dense bodies in all five patients in our cohort who had adequately preserved samples for electron microscopic examination suggests that this finding may be a sensitive diagnostic feature of ABCA3 mutations. The absence of mature lamellar bodies and accumulation of these central or eccentrically placed dense bodies have previously been reported in patients with ABCA3 mutations.3 10 13 16–18 In three of our five patients some normal lamellar bodies were also seen in type II alveolar cells, which previously had been described in only one patient.10 This finding further underscores the variability in the histology that can occur with ABCA3 mutations. More importantly, however, given that results of DNA analyses are not quickly available, emphasis must be placed on the need to preserve a portion of any open lung biopsy done for evaluation of chILD for electron microscopy analysis, as it may provide a rapid preliminary diagnosis of ABCA3 mutations. Guidelines for the processing of paediatric lung biopsies have recently been published.19
The significant heterogeneity and number of novel mutations in the ABCA3 gene found in our patients is similar to the report by Brasch et al10 and suggests that, like other genetic conditions (eg, cystic fibrosis), many more mutations may yet be discovered. The E292V mutation found in two of our patients has been reported as the most common defect in the ABCA3 gene leading to chILD.20 Our study shows that the outcome in children with ABCA3 mutations is also quite variable. Almost half of our patients have survived into the second decade of life without need for lung transplantation. However, we realise that a selection bias may exist in our study due to the referral pattern.
The clinical and radiological features described above for ABCA3 mutations can overlap with those seen with other forms of inborn errors of surfactant metabolism (SP-B and SP-C mutations). Respiratory distress syndrome in term newborns can result from either SP-B or ABCA3 mutations,1 while chronic ILD starting in infancy or early childhood can be the manifestation of either SP-C or ABCA3 mutations.2 4 5 7 21–24 Young children with ILD due to SP-C mutations can have similar findings on CT imaging as those with ABCA3 mutations.7 21 22 While histological findings can also overlap,1 4–7 25 analysis of the lung ultrastructure using electron microscopy may be able to distinguish between these three entities. Patients with SP-B mutations have large, membrane-bound, pleomorphic, cytoplasmic inclusions with irregularly disposed lamellae and multiple associated vesicular structures,17 while patients with SP-C mutations can have normal or (less commonly) somewhat disorganised-appearing lamellar bodies.4 23 24
Based on the results of our analysis, we conclude that ABCA3 mutations should be considered in the differential diagnoses of newborns with unexplained respiratory distress syndrome and for older infants and young children who have chronic ILD of unclear aetiology. Findings on HRCT scanning—particularly widespread ground-glass opacification, septal thickening and parenchymal lung cysts—can provide supportive evidence of a possible inborn error in surfactant metabolism. DNA analysis for mutations in the ABCA3 gene is necessary for a definitive diagnosis and should also be considered for the SP-B and SP-C genes because of similarities in the clinical, radiological and pathological features of these entities. In patients with suspected ABCA3 mutations and chronic stable disease, confirmatory genetic testing will eliminate the need for a lung biopsy. If genetic testing is equivocal or non-diagnostic, a lung biopsy specimen should be obtained to look for one of the compatible histological patterns and to evaluate for characteristic lamellar dense bodies by electron microscopy. A lung biopsy should also be considered in patients with suspected ABCA3 mutations and rapidly progressive disease. These patients could be candidates for lung transplantation, and awaiting genetic testing results would delay a timely diagnosis. Outcomes in patients with ABCA3 mutations are variable, ranging from severe irreversible respiratory failure in early infancy to chronic static or progressive interstitial lung disease, with many patients surviving well into their second decade without lung transplantation.
Acknowledgments
The authors thank Drs Phil Black, Fran White, Stuart Sweet, Susan Wert, Gail Deutsch, Lisa Young, Aaron Hamvas, Robert Zwerdling, Jason Fullmer and Tarak Patel for their care of these patients and their assistance in our research. We recognise Mr Jim Barrish for his technical expertise in electron microscopy. We also express our appreciation to the patients and their families for their assistance and willingness to participate in the study.
REFERENCES
Footnotes
Funding: LMN was supported by an NIH grant (HL-54703).
Competing interests: None.
Ethics approval: This study was approved by the Institutional Review Board of Baylor College of Medicine.