Article Text

Abstract
Background: Acute lung injury is an important cause of respiratory failure in the critically ill patient. It is caused by damage to the alveolar barrier with subsequent alveolar flooding leading to the development of refractory hypoxaemia. β Agonists stimulate alveolar fluid clearance in animal models of lung injury. In a clinical trial (BALTI-1), intravenous beta agonists reduced extravascular lung water, an effect that took 72 h in contrast with what animal studies suggest. One possible explanation for the delay in change in extravascular lung water is the time required for salbutamol to stimulate alveolar epithelial repair.
Objective: To investigate whether salbutamol can stimulate alveolar epithelial repair in vivo and in vitro.
Results: Intravenous salbutamol reduced measures of alveolar–capillary permeability in patients with acute respiratory distress syndrome (ARDS). In vitro, salbutamol stimulated both wound repair, and spreading and proliferation of A549 cells and distal lung epithelial cells. Lung lavage fluid from patients treated with salbutamol enhanced wound repair responses compared with placebo treated patients in vitro by an interleukin 1β dependent mechanism.
Conclusions: Our in vivo and in vitro work suggests that salbutamol may stimulate epithelial repair—potentially a pharmacological first in ARDS. Clearly establishing the mechanisms and pathways responsible for this is important for the future, and may allow identification of novel therapeutic targets to promote alveolar epithelial repair in humans with ARDS.
Statistics from Altmetric.com
Acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) are important causes of respiratory failure in the critically ill patient.1 Pathologically, ARDS is characterised by intense inflammation in the alveolar space, with extensive physical damage to the alveolar–capillary membrane. This leads to the outpouring of proteinacious fluid from the pulmonary microcirculation causing alveolar flooding and the development of non-cardiogenic pulmonary oedema. There is considerable heterogeneity in the subsequent course of ARDS. Some patients demonstrate rapid repair of the alveolar–capillary barrier while others enter a more chronic phase of the condition. The presence of an intact epithelial barrier has been shown to be associated with improved outcomes in patients with ARDS.2 Treatments aimed at improving epithelial function might therefore become one of the key elements to accelerate recovery and decrease the mortality of patients with ARDS.3
β2 Agonists accelerate alveolar fluid clearance within a matter of minutes to hours in in vitro experimental studies,4 animal models of ARDS5 and ex vivo human lung.6 In a randomised, double blind, placebo controlled clinical trail (the Beta Agonist Lung Injury TrIal (BALTI-1)), we demonstrated that 7 days of treatment with 15 μg/kg/h of intravenous salbutamol reduced extravascular lung water in patients with ARDS.7 In contrast with our original hypothesis, the effect was not immediately apparent but delayed until 72 h after initiation of treatment.
Several different lines of research suggest that β agonists may promote alveolar epithelial repair. In mice, β adrenergic tone has a protective effect on lung fluid balance in acute bacterial pneumonia.8 This occurred at least in part as a result of reduced alveolar capillary permeability. In vitro, isoproterenol stimulates the closure of mechanically induced wounds of monolayers of bovine bronchial epithelial cells by increasing cAMP and activating protein kinase A (PKA).9 Thus one possible explanation for the delay in change in extravascular lung water is the time required for salbutamol to stimulate alveolar epithelial repair. The aim of the present study therefore was to investigate whether salbutamol can stimulate alveolar epithelial repair.
METHODS
Clinical study
Adult patients undergoing mechanical ventilation enrolled in a randomised controlled trial (BALTI-1) investigating the effects of intravenous salbutamol on extravascular lung water within 48 h of onset of ALI and ARDS were eligible for inclusion in this study.7 ALI and ARDS were defined according to the definition of the American European Consensus Conference.10 Exclusion criteria were: age <18 years; participation in other intervention trials; severe obstructive airways disease requiring nebulised or intravenous β2 agonist; treatment with β blockers within 48 h; neutrophil count <0.3×109 l; brainstem death; treatment withdrawal within 24 h; immunosuppression (steroids >20 mg/day, chemotherapy or other immunosuppressive agents within 2 weeks); lobectomy/pneumonectomy; burns >40% body surface area; assent declined from the next of kin.
Biological sample collection and processing
Bronchoalveolar lavage (BAL), using 150 ml of cold saline, was performed immediately following randomisation and whenever possible 4 days later. Lavage fluid was kept on ice until transferred to the laboratory where it was filtered through course surgical gauze to remove mucous and other debris. The fluid was then spun at 500 g for 5 min in a prechilled centrifuge (4°C). The supernatant was removed and immediately frozen to −80°C and stored for subsequent analysis.
Patient demographics and scoring
Patient demographic characteristics were recorded at baseline. The Acute Physiology and Chronic Health Evaluation II (APACHE II) and Simplified Acute Physiology Score II (SAPS II) score, predicted ICU mortality were recorded as global markers of disease severity.11 The Murray Lung Injury Score12 and arterial oxygen tension:fractional inspired oxygen (Pao2:Fio2) ratio were collected as markers of the severity of lung injury.
Alveolar capillary permeability
The single indicator transpulmonary thermodilution system (PiCCO; Pulsion Medical Systems, Munich, Germany) was used to calculate an in vivo alveolar–capillary permeability index. The permeability index was derived from the ratio of extravascular lung water divided by pulmonary blood volume. Previous studies have shown that this index can separate cardiogenic (low permeability) and inflammatory (high permeability) causes of pulmonary oedema.13 14 Protein selectivity in lavage fluid was used as a second measure of alveolar capillary permeability15 by measuring the ratio of IgG:total protein. IgG was measured by polyethylene glycol enhanced immunoturbidimetry (DakoCytomation, Ely, UK) and total protein by the Lowry method.16
Cell culture
Primary cultures of human distal lung epithelial cells (DLEC) (Cambrex, San Diego, California, USA) were cultured in complete growth media (SAGM; Cambrex) according to the manufacturer’s protocol. DLEC are non-ciliated primary adult lung epithelial cells isolated from distal airways of less than 1.0 mm in diameter. These cells grow as flat monolayers, stain positive for cytokeratin and surfactant protein A, and show lamellar body-like structures on electron microscopy. They do not contain neurosecretory granules by electron microscopy.
The cells were seeded in 25 cm2 flasks at a density of 2500 cells/cm2 and incubated at 37°C with 5% CO2. Cells were used for the experiments after the first or second subculture. Under these culture conditions, cells form monolayers and do not differentiate into ciliated cells.17 Cells were obtained from three separate donors and all experiments were repeated in triplicate before passage 3. A549 human epithelial cells (ECACC; Sigma UK) were similarly cultured in DMEM culture media supplemented with F-10 nutrient mixture (Sigma) at 37°C with 5% CO2. Cells were subcultured at 60–80% confluence using trypsin/EDTA.
Epithelial wound repair
Epithelial repair was determined using an in vitro epithelial wound repair assay, as described previously.17 18 Briefly, A549 alveolar epithelial-like cells were cultured to confluence in 24 well plates in DMEM containing 10% fetal bovine serum and then mechanically wounded with a pipette tip. In addition, primary human distal lung epithelial cells (Cambrex) were cultured in complete growth media (SAGM; Cambrex) according to the manufacturer’s protocol before wounding with a pipette tip. Both A549 epithelial cells and primary lung distal epithelial cells were plated on untreated culture plates. Preliminary experiments demonstrated maximal wound repair at 24 h. After wounding, fresh control media, salbutamol (10−6 M) (GlaxoSmithKline, UK) or a 50:50 mix of BAL fluid/culture media were added to the wounded alveolar epithelial monolayers and the area of the denuded surface was measured immediately and again after 24 h
The plates were placed on an inverted microscope (Axiovert 35; Zeiss, Thornwood, New York, USA), and the cell monolayer was photographed with a digital camera (C 2400; NEC, Hawthorne, California, USA) connected to the microscope. The image was later captured by an image analysing frame-grabber card (LG-3 Scientific Frame Grabber; Scion, Frederick, Maryland, USA) and analysed with image analysis software (NIH Image 1.55). Repair is expressed as the percentage of the original wound area covered by cells. Subsequent to photography, monolayers were incubated with 100 μl of cell titre (see below) to assess relative cell counts between the monolayers. In separate experiments, to assess cell spreading, monolayers were stained with diff-quick (Fisher Scientific, UK) and images obtained of the wound at 24 h. Cell spreading was measured as the inter-nuclear distances measured at fixed points on the image.19 To measure the contribution of bioactive interleukin 1β (IL1β) and vascular endothelial growth factor (VEGF) in BAL fluid, specific inhibitors were added to the wound repair model (interleukin 1 receptor antagonist 200 ng/ml (R&D systems, Abingdon, UK) and sVEGFR-1 25 ng/ml (R&D Systems)).
Cell proliferation assay
After 24 h of culture, cell proliferation was assessed by adding 20 μl of CellTiter 96 AQueous one solution cell proliferation solution (Promega, UK) to 100 μl of cell culture media in a 96 well culture plate (Nunc, Paisley, UK) according to manufacturer’s instructions. The reaction was allowed to proceed for 2 h at 37°C, in an atmosphere containing 5% CO2. The cell titre solution contains a tetrazolium compound which is metabolised by healthy proliferating cells to a formazan product the absorbance of which was read at 495 nm on an MRX-II 96 well plate reader (Dynex Technologies, UK). Data from proliferation bioassays comparing the CellTiter 96 AQueous Assay and [3H]-thymidine incorporation show similar results.20 Furthermore, in preliminary experiments, there was a linear relationship between cell titre readings and proliferation, as measured by BRDU incorporation (Calbiochem, UK) over a range of cell counts of 2500–40000 cells per well (r = 0.97, p = 0.001, data not shown).
VEGF and IL1β ELISA
Immunoreactive VEGF and IL1β were measured by commercial ELISA (R&D Systems). The concentration of VEGF and IL1β in the epithelial lining fluid was calculated using the urea dilution method.21
Reagents
Salbutamol (GlaxoSmithKline, UK) was diluted in appropriate media to 10−6 M. This dose was used as it represents the physiological level of salbutamol achieved in the plasma of ARDS patients in the BALTI-1 study.22 Other reagents used were propranolol (Sigma, UK) 10−5 M and IL1 (R&D Systems).
Statistical methods
The study was powered to detect a 15% difference in wound repair between the placebo and salbutamol groups based on the data from Geiser et al who examined wound repair rates with pulmonary oedema fluid.19 We calculated that nine patients would need to be recruited in each arm to detect this difference with 80% power at a significance level of 0.05. Statistical analysis was done using the Student’s unpaired t test, paired t test or ANOVA, when appropriate. If ANOVA revealed a significant difference between groups, it was followed by a post hoc Tukey’s test. Linear associations were tested using Pearson’s correlation test. Data are expressed as mean (SD) unless otherwise stated; p<0.05 was considered significant.
RESULTS
Patient demographics
Forty patients were enrolled in the BALTI-1 and of these 19 received salbutamol and 21 placebo. Twenty-two patients had BAL at baseline and on day 4 and are included in this study (nine salbutamol, 13 placebo). Patient demographics, and severity and aetiology of lung injury for this subgroup are presented in table 1. Patient demographic and clinical data for the full BALTI patient population have been published previously.7
Treatment with salbutamol reduces measures of alveolar–capillary permeability in vivo
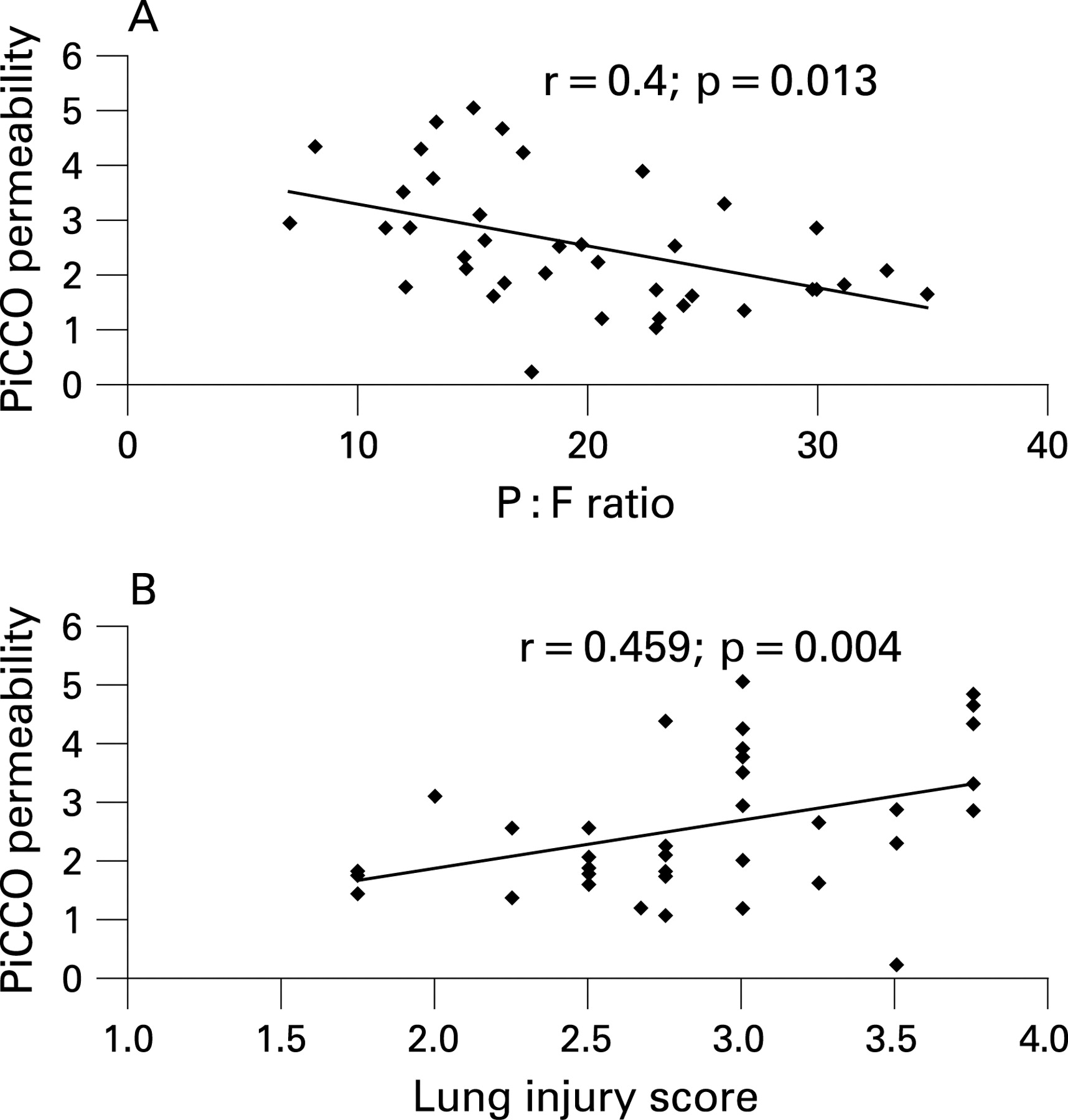
Alveolar–capillary permeability was measured in vivo using the PiCCO permeability index. The permeability index correlated with both lung injury score (r = 0.4, p = 0.013) and severity of hypoxaemia (r = 0.459, p = 0.004) (fig 1), suggesting it is associated with the severity of alveolar damage.
There were no significant differences in permeability index between the salbutamol and placebo groups at baseline. However, the permeability index was significantly reduced in the salbutamol treated group compared with placebo at day 4 (0.6 (0.1) vs 1.6 (1.2), 95% CI 0.2 to 1.7; p = 0.02) and day 7 (0.5 (0.2) vs 1.9 (2.0), 95% CI 0.1 to 2.7; p = 0.035) (fig 2).

Early during ARDS, physical damage to the epithelium results in alveolar flooding with plasma proteins. The size selectivity of the epithelial barrier increases allowing large molecular weight proteins such as IgG (150 000 Da) into the alveolar space.15 Thus estimation of these proteins relative to total protein allows an estimate of the size selectivity of the epithelial barrier. We therefore investigated the ratio of IgG to total protein in BAL fluid. There was no difference in IgG:total protein ratio at baseline or at 24 h. However on day 4, the ratio of IgG:total protein was significantly reduced in the salbutamol treated group (0.5 (0.06) vs 1.1 (0.8), 95% CI 0.01 to 0.1; p = 0.021) suggesting there was a reduction in alveolar capillary permeability in vivo (fig 3). There was no significant difference in total protein at any of the time points (data shown in fig 3).

Salbutamol promotes in vitro alveolar wound repair
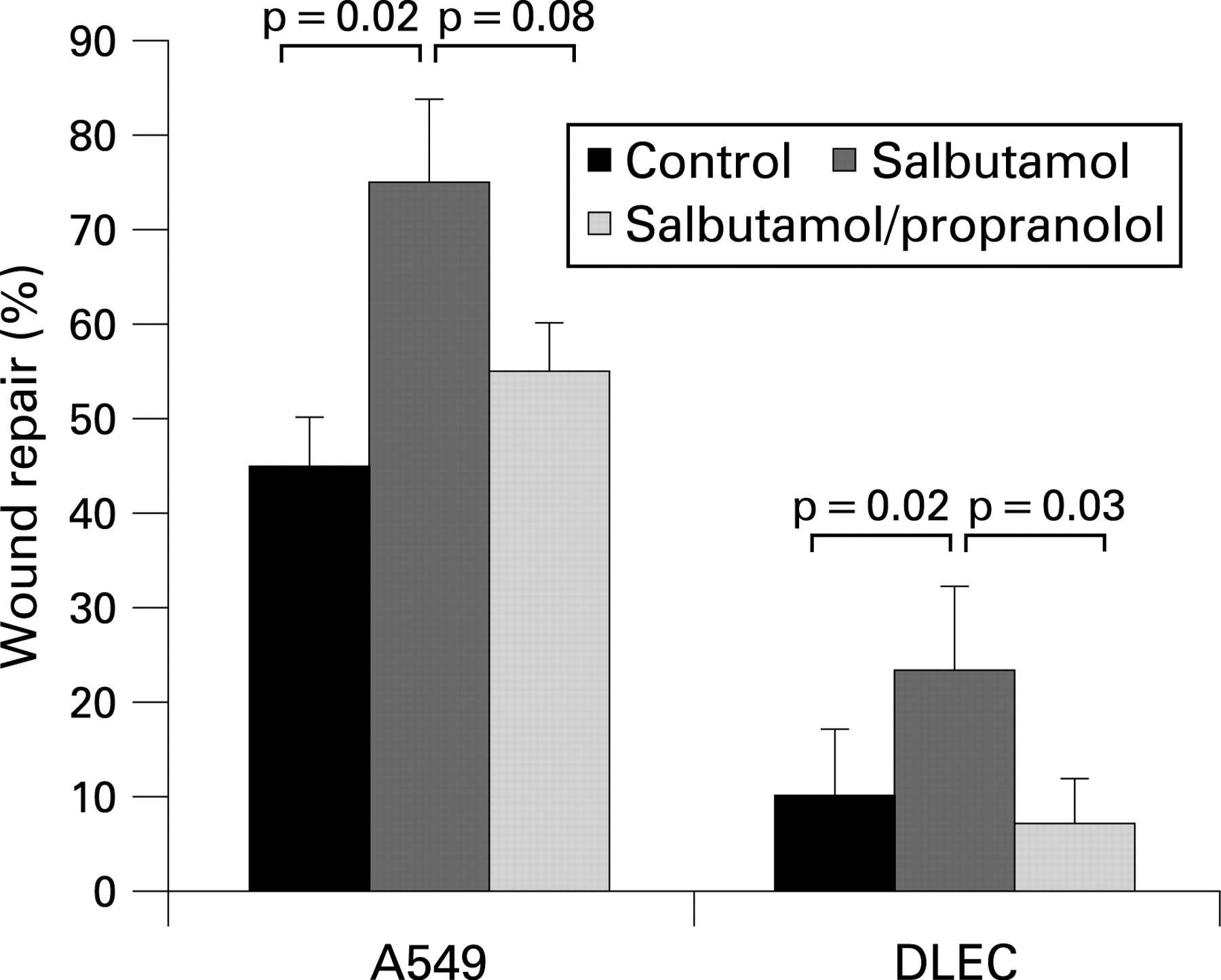
A prerequisite for the hypothesis that salbutamol stimulates epithelial repair is to demonstrate in vitro that physiological concentrations of salbutamol have these effects on human lung epithelial cells. Treatment of wounded A549 monolayers with 10−6 M salbutamol significantly increased wound repair (mean wound% closure salbutamol 75.0 (4.1)% vs control 44.7 (7.1)%, 95% CI 16% to 43%; p = 0.003). This effect was blocked by co-incubation with 10−5 M propranolol (55.5 (4.6)%; p = 0.008 compared with salbutamol). Enhanced wound repair was also seen when wounded DLEC monolayers were incubated with 10−6 M salbutamol (wound closure salbutamol 26.1 (7.1)% vs control 10.1 (8.2)%, 95% CI 0.3% to 31.8%; p = 0.047) (fig 4).
Salbutamol promotes wound repair through proliferation and cell spreading
Wound repair may occur due to cellular spreading and/or proliferation. To establish if salbutamol stimulated epithelial cell proliferation, A549 and DLEC cells were incubated for 24 h in control media with and without salbutamol 10−6 M. Salbutamol increased A549 and DLEC cell proliferation by 18% (p = 0.015) and 25% compared with controls (p = 0.012), respectively. Inter-nuclear distances also increased in DLEC in response to stimulating wounds with salbutamol (control 54.4 (1.9) vs salbutamol 58.9 (1.6); p = 0.045) suggestive of increased cell spreading in the salbutamol treated monolayers.
A role for VEGF in the alveolar repair response to salbutamol?
Alveolar type II cells are the predominant source of VEGF in the lung.23 Previous studies have shown that an increase in alveolar VEGF early in the course of ARDS is associated with resolution of lung injury. VEGF is a potent stimulus for endothelial and epithelial repair.24 25 We therefore measured VEGF in the BAL fluid of the BALTI patients and compared epithelial lining fluid (ELF) levels of treated and untreated patients as a marker of alveolar repair.26 BAL fluid levels of VEGF did not differ between treated and untreated patients on day 4. ELF VEGF levels, however, were significantly greater in the salbutamol treated patient (10.2 (6.5) vs placebo 5.3 (2.7), 95% CI 0.3 to 0.9) ng/ml ELF), providing supporting evidence that salbutamol treatment was associated with alveolar repair. The effect of salbutamol does not appear to be due to increased epithelial cell production of VEGF since salbutamol did not stimulate production of VEGF from A549 and DLEC cells in vitro (data not shown). Furthermore, blocking experiments with sVEGR-1 did not alter the rate of epithelial wound closure (data not shown) suggesting that VEGF made little contribution to the epithelial response to BAL fluid in our wound repair model.
BAL fluid from patients with ARDS treated with salbutamol promotes wound repair and is IL1β dependent
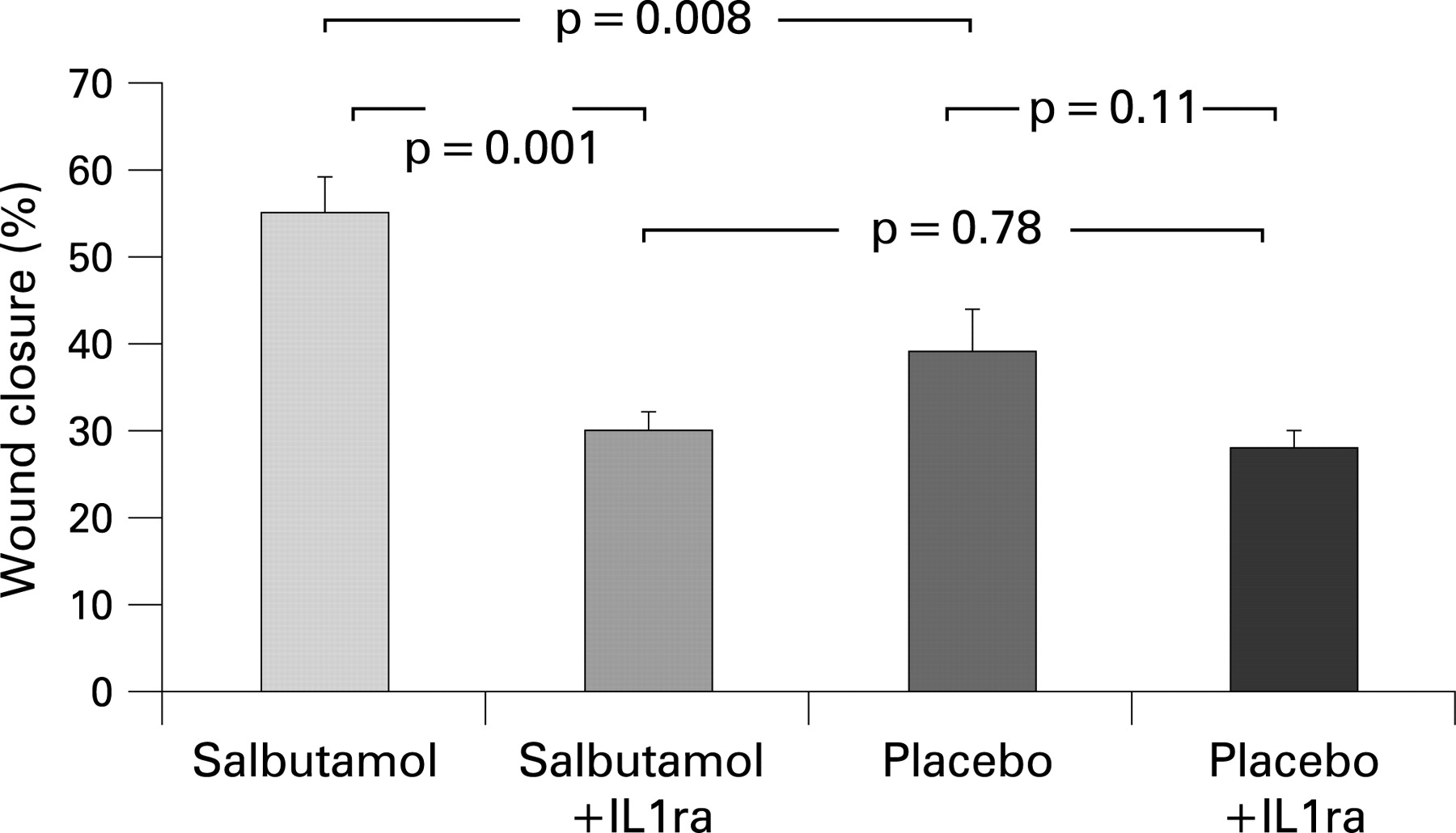
Compared with saline controls, incubation of the wounded monolayer with BAL fluid from patients at the onset of ARDS significantly increased wound repair (BAL fluid 40.1 (4)% vs saline control 20.2 (3)%; p = 0.001). The rate of wound closure following incubation of BAL fluid from patients after 4 days of treatment with intravenous salbutamol was significantly greater than for placebo treated patients (salbutamol 52.7 (4.4)% vs placebo 46.2 (4.3)%, respectively; p = 0.008) (fig 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Previous studies of oedema fluid from ARDS patients have implicated IL1β in the wound repair response.19 27 Although there was no significant difference in the amount of immunoreactive IL1β measured by ELISA in BAL fluid between the two groups (salbutamol 143.7 (266) vs placebo 178.9 (222) pg/ml; p = 0.7; 95% CI −278 to 208 pg/ml), blocking experiments with IL1β antagonist significantly reduced wound repair in the salbutamol treated group (data shown in fig 5).
DISCUSSION
Experimental studies have shown that β2 agonists rapidly accelerate alveolar fluid clearance in in vitro and in animal models of lung injury.28 We tested the hypothesis that treatment with an intravenous infusion of salbutamol in patients with ARDS would reduce lung water.7 Although we observed a significant reduction in lung water in the salbutamol group, this effect was not evident until 72 h after initiation of treatment. We hypothesised that failure to see an early response may have been due to extensive damage to the alveolar–capillary membrane, leading to ongoing alveolar flooding which exceeded the capacity of fluid clearance mechanisms. This study set out to investigate the effects of salbutamol on alveolar–capillary permeability and repair.
The principle findings of this study are that intravenous salbutamol reduced measures of alveolar–capillary permeability in ARDS. This suggests that treatment with β2 agonists may also have an effect on repair of the alveolar capillary barrier. As VEGF is produced predominantly by type II cells in the lung, and has been suggested as a marker of alveolar recovery,26 our finding of increased epithelial lining fluid VEGF levels in the salbutamol treated group further supports this hypothesis.
The alveolar capillary barrier comprises two discrete layers—the alveolar epithelium and pulmonary capillary endothelium. There are several putative mechanisms through which β2 agonists may enhance recovery from ARDS. Firstly, β2 agonists may reduce the severity of damage to the alveolar–capillary barrier. Experimental studies show that β2 agonists can modulate the inflammatory cascade thereby reducing ongoing damage to the alveolar capillary barrier.28 However, in BALTI-1, there were no differences in neutrophil recruitment, activation or inflammatory cytokine (IL8, tumour necrosis factor α) production in our patients.22 β2 Agonists may also protect against infection induced epithelial damage. Salmeterol, a long acting β2 agonist, reduced Pseudomonas aeruginosa and Haemophilus influenzae induced epithelial cell damage in vitro, probably by maintaining intracellular cAMP concentrations, which together with adenosine triphosphate are known to fall under these conditions.29 30 Treatment of our patients with salbutamol was also associated with a significant reduction in plateau airway pressures7 which may have reduced the magnitude of ventilation associated epithelial and endothelial injury.
Alternatively, β2 agonists could enhance repair to the alveolar–epithelial barrier through effects on endothelial or epithelial cells. Previously, β2 agonists have been shown to reduce endothelial permeability in acute lung injury in sheep31 and rats.5 32 This is thought to be due to increasing the intracellular concentration of cAMP, which relaxes actin–myosin contraction. Relaxation of the endothelial cytoskeleton increases cell–cell contact, cell surface area and apposition, thereby improving endothelial barrier function.33 These experimental findings are supported by a small non-randomised study in humans where administration of intravenous terbutaline to 10 patients with ARDS was associated with a significant reduction in lung vascular permeability (measured by radiolabelled transferrin) and improved survival.34
Early epithelial repair with re-epithelialisation of the denuded basement membrane is important for the restoration of the normal architecture of the lung. Effective alveolar repair after ARDS is believed to involve the transdifferentiation of alveolar type II cells which retain stem cell-like properties into type I cells via intermediate cell phenotypes.35 The turnover rate of ATII cells is boosted after acute lung injury36 37 and the recovery process is believed to involve cell migration and proliferation in addition to transdifferentiation of ATII epithelial cells.
ARDS is characterised by intense inflammation within the alveolar space with evidence of increased bioactive IL1β early after the onset of ARDS.38 Geiser et al were the first to show that pulmonary oedema fluid, early in the course of ARDS, stimulates repair of wounded monolayers in culture to a greater extent than plasma obtained from the same patients or pulmonary oedema fluid from patients with hydrostatic oedema.19 The potential of oedema fluid to promote wound repair was associated with a trend towards improved survival and reduction in the duration of ventilation.27 The enhanced wound repair was IL1β dependent and mediated by autocrine release of epidermal growth factor and transforming growth factor α. In our study, BAL fluid similarly stimulated epithelial wound repair in vitro. Moreover, BAL fluid from patients treated with intravenous salbutamol for 4 days stimulated significantly greater wound repair than BAL fluid from the placebo arm. This effect was abrogated by anti-IL1β.
Monocytes and macrophages are thought to be the predominant source of IL1β in ARDS, although neutrophils, endothelial cells and type II epithelial cells are also likely to contribute to the increased levels observed in ARDS. Previous studies have produced conflicting results with respect to the effect of β2 agonists on IL1β production. Some studies suggest suppression of inflammatory cytokine production in whole blood39 while others report increased transcription and release of IL1β and other inflammatory cytokines.40 In this study, immunoreactive IL1β was not different between the treated and placebo patients at day 4. This suggests perhaps that other factors within the BAL fluid of the treated patients may act in a synergistic cytokine network to augment the bioactivity of IL1β in the salbutamol treated patients..
It is also possible that β2 agonists may have a direct effect on epithelial repair. In the BALTI-1 study, treatment with intravenous salbutamol at 15 μg/kg/h achieved circulating concentrations of salbutamol of 10−6 M,22 similar to levels found in the alveolar space following nebulised salbutamol by Atabai et al.41 Our in vitro studies using physiological concentrations of salbutamol enhanced epithelial cell proliferation and cell spreading. The cellular regulatory mechanisms necessary for coordinating functional repopulation and reconstitution of the alveolar–capillary membrane remain unclear but may be controlled in part by regulatory pathways that are important in lung morphogenesis and development. The Wnt/β-catenin pathway has been identified as one of the numerous signalling pathways critical for precise temporal and spatial control of lung morphogenesis.42 Over expression of β-catenin in mice leads to elevated epithelial cell differentiation and surfactant expression.43 β Agonists might interact with the Wnt/β-catenin pathway as cAMP analogues and isoproterenol increase the cytoplasmic and nuclear β-catenin protein level via PKA.44 As PKA inhibits the ubiquitination of β-catenin by phosphorylating β-catenin, β2 agonist activation of PKA may cause β-catenin to accumulate and thus promote an alveolar epithelial repair response. Interestingly, IL1β has also been reported to positively regulate the WNT/β-catenin pathway via nuclear factor κB activation, suggesting that there could potentially be intracellular molecular synergy between the actions of IL1β and salbutamol in the wound repair response that is worthy of further study.45
There are some limitations to this study. Firstly, the in vivo PiCCO permeability index has not been fully validated in humans with ARDS. Although in animal models it can differentiate between cardiogenic and non-cardiogenic pulmonary oedema,13 only limited studies in humans have confirmed the utility of this index.14 However, the parallel findings of a reduction in selective protein permeability supports the PiCCO derived observations. Secondly, our in vitro models do not completely represent the in vivo alveolar environment. BAL fluid was diluted in a 50:50 mix with cell culture media which has previously been reported as the optimal dilution for this experimental model.19 46 Epithelial cells were grown in isolated monolayers, rather than in contact with extracellular matrix and endothelial/mesenchymal cells. It is possible that the repair activity is different in vivo when compared with the ex vivo conditions. Thirdly, we used the human cell lines A549 and primary distal lung epithelial cells for our studies and not primary human alveolar type II epithelial cells. Although these cell lines have been used in many studies, they may not respond to all biological stimuli in the same fashion as primary alveolar epithelial type II cells.
In conclusion, our in vivo and in vitro work suggests that salbutamol may stimulate epithelial repair, potentially a pharmacological first in ARDS. Clearly establishing the mechanisms and pathways responsible for this is important for the future, and may allow identification of novel therapeutic targets to promote alveolar epithelial repair in humans with ARDS.
Acknowledgments
The authors would like to thank Dr Peter Gosling and Miss Sophie Mahon for assistance with laboratory assays.
REFERENCES
Footnotes
-
Funding: GDP is supported by a DH (NIHR) Clinician Scientist Award. DRT is supported by the Wellcome Trust.
-
Competing interests: DT and GDP have received honoraria for speaker meetings and educational grants to attend conferences from manufacturers of beta agonists. The are no other competing interests.
-
Ethics approval: The study was approved by the local research ethics committee.
Linked Articles
- Airwaves