Article Text

Abstract
Background: Pneumocystis pneumonia (PCP) is conventionally diagnosed by identifying Pneumocystis jirovecii in lower respiratory tract samples using cytochemical stains. Molecular diagnosis of PCP is potentially more sensitive.
Methods: A study was undertaken to use an extensively optimised real-time polymerase chain reaction (PCR) using primers designed to hybridise with the P jirovecii heat shock protein 70 (HSP70) gene to quantify P jirovecii DNA in bronchoalveolar lavage (BAL) fluid from HIV-infected patients with and without PCP, and to compare this assay with conventional PCR targeting the P jirovecii mitochondrial large subunit rRNA gene sequence (mt LSU rRNA).
Results: Sixty-one patients had 62 episodes of PCP (defined by detection of P jirovecii in BAL fluid by cytochemical stains and typical clinical presentation). Quantifiable HSP70 DNA was detected in 61/62 (range ∼13–18 608 copies/reaction; median ∼332) and was detectable but below the limit of quantification (∼5 copies/reaction) in 1/62. Seventy-one other patients had 74 episodes with alternative diagnoses. Quantifiable HSP70 DNA was detectable in 6/74 (8%) episodes (range ∼6–590 copies/reaction; median ∼14) and detectable but below the limit of quantification in 34/74 (46%). Receiver-operator curve analysis (cut-off >10 copies/reaction) showed a clinical sensitivity of 98% (95% 91% to 100%) and specificity of 96% (95% CI 87% to 99%) for diagnosis of PCP. By contrast, clinical sensitivity of mt LSU rRNA PCR was 97% (95% CI 89% to 99%) and specificity was 68% (95% CI 56% to 78%).
Conclusion: The HSP70 real-time PCR assay detects P jirovecii DNA in BAL fluid and may have a diagnostic application. Quantification of P jirovecii DNA by real-time PCR may also discriminate between colonisation with P jirovecii and infection.
Statistics from Altmetric.com
The fungal pathogen Pneumocystis jirovecii is the cause of Pneumocystis pneumonia (PCP) in humans.1 Diagnosis of P jirovecii infection is hampered by the lack of a sustainable in vitro culture method. Diagnosis of PCP is typically made by direct examination of respiratory samples (bronchoalveolar lavage (BAL) fluid or induced sputum) after staining in order to detect the cyst form of Pneumocystis.2 Some laboratories also use immunofluorescence stains to enhance sensitivity. The use of cytochemical stains for diagnosis is time consuming, and it may be difficult to maintain laboratory diagnostic expertise because of the lower incidence of PCP since the introduction of highly active antiretroviral therapy.
Molecular diagnostic techniques using the polymerase chain reaction (PCR) are more clinically sensitive than staining for detection of P jirovecii in BAL fluid and induced sputum.3–6 Molecular techniques, however, may identify P jirovecii DNA in respiratory samples from patients without clinically apparent PCP,7–10 suggesting asymptomatic carriage or “colonisation”. Several studies have used real-time PCR for detection of P carinii,11 P murina12 and P jirovecii in respiratory specimens.13–21
Real-time PCR allows accurate quantification of DNA and the potential to discriminate between asymptomatic carriage of P jirovecii and clinical disease based on pathogen load. The objective of this study was to compare real-time PCR using primers designed to hybridise to the heat shock protein 70 (HSP70) gene of P jirovecii22 23 with conventional PCR using primers designed to the large subunit of mitochondrial rRNA (mt LSU rRNA)3 4 10 for detection of P jirovecii DNA in BAL fluid from patients undergoing diagnostic bronchoscopy. Patients had either proven PCP or confirmed alternative diagnoses.
METHODS
Selection of a new molecular target
The target region of HSP70 was obtained for P jirovecii by designing generic primers to the flanking regions of P carinii. Nucleotide sequences for all Pneumocystis types available from public data repositories were searched using BlastX24 against the non-redundant protein database. Expressed sequence tag data were assembled into contigs using CAP3 prior to searching in order to remove redundancy and improve sequence accuracy. Sequences with high quality matches (bit score >60) to proteins from a broad spectrum of eukaryotic species were considered further (minimally: Caenorhabditis elegans, Drosophila melanogaster, Saccharomyces cerevisiae, Schizosaccharomyces pombe and Neurospora crassa).
The corresponding region was amplified using P jirovecii as template and sequenced. Real-time PCR for P jirovecii HSP70 (PjHSP70a) was designed to amplify a 106 base pair region of the P jirovecii sequence and not the corresponding sequence of other Pneumocystis species, or a wide range of potential fungal, bacterial or mycobacterial pathogens (table 1). DNA was extracted from these isolates using the DNeasy tissue kit (QIAGEN, Crawley, UK) following the manufacturer’s instructions, and was used as template for the PjHSP70a.
Patients and samples
One hundred and thirty-six BAL samples were obtained from 132 adult HIV-infected patients (112 men) undergoing diagnostic bronchoscopy. Sixty-one consecutive patients with PCP had 62 episodes of PCP; one patient had two episodes (interval 9 weeks). All had typical clinical and radiographic presentations, identification of P jirovecii cysts in BAL fluid by Grocott-Gomori methenamine silver staining2 and response to specific anti-PCP therapy.25 26 A further 71 consecutive patients (74 episodes) investigated contemporaneously did not have PCP clinically or radiographically, had negative results from Grocott-Gomori staining and did not receive specific anti-Pneumocystis treatment. All had alternative diagnoses, as described previously,10 26–28 comprising pulmonary Kaposi sarcoma (n = 21; one patient had two BAL, interval 27 weeks), bacterial bronchitis (n = 12), lymphocytic interstitial pneumonitis (n = 5; one had two BAL, interval 24 weeks), Cryptococcus neoformans pneumonia (n = 3), pulmonary tuberculosis (n = 3), Aspergillus fumigatus bronchitis (n = 3; one had two BAL, interval 3 weeks, the diagnosis then was bacterial bronchitis), S pneumoniae pneumonia (n = 2), bronchiectasis (n = 2), self-limiting fever and dyspnoea (n = 2). One patient each had cytomegalovirus pneumonitis, pulmonary Mycobacterium avium infection, Salmonella typhi septicaemia, tracheitis, adenocarcinoma of bronchus, primary effusion lymphoma, pulmonary Castleman disease and HIV “constitutional” disease. No patient with an alternative diagnosis developed PCP during the 6-month follow-up period.
Outcome (survival at 1 month) was recorded in all patients. Additionally, in those with PCP, receipt of specific anti-Pneumocystis prophylaxis, disease severity (arterial oxygen tension (Pao2) breathing room air) and number of days of specific anti-Pneumocystis treatment before BAL were also recorded. All bronchoscopies were performed by one of the authors (RFM); BAL was performed in a standardised manner as previously described.25 26 All BAL samples were coded and analyses by PCR were performed “blind” to the patient’s clinical and laboratory details. All patients undergoing bronchoscopy gave informed written consent and the study was performed with approval from the local research ethics committee.
DNA extraction
DNA extraction from BAL fluid was first done using the DNeasy tissue kit and subsequently using the QIAamp UltraSens Virus Kit (both QIAGEN, Crawley, UK) following the manufacturer’s instructions. Total DNA was extracted from 200 μl (DNeasy) or 750 μl (QIAamp) BAL fluid. RNA quench was also included in the latter extract, as suggested by the manufacturer, and the extracted sample was eluted in 60 μl of elution buffer by contrast with the DNeasy extraction (100 μl of elution buffer). QIAamp UltraSens Virus Kit was chosen as it was specifically designed to extract DNA from liquid samples, enabled a greater volume to be extracted from BAL fluid and facilitated generation of a higher concentration of extracted nucleic acids. Consequently, the final QIAamp eluate achieved a 12.5-fold concentration compared with a twofold increase with the DNeasy kit. When the two extraction techniques were both applied to BAL samples, a greater amount of DNA was extracted with the QIAamp method than with the DNeasy method (fig 1).

Molecular assessment of P jirovecii DNA in BAL fluid
Attempts were made to use the mt LSU rRNA assay by real-time PCR, but efficient amplification was not possible and so conventional PCR was used for comparison with the PjHSP70a assay.
PjHSP70a
The PjHSP70a assay consisted of a 12.5 μl reaction using 1 U Hot Taq (BioGene, Kimbolton, UK) with supplied buffer, 3 mM MgCl2, 200 μM each dNTP, 250 μg/μl tRNA (Sigma, UK) and 300 nM of the forward (5′-CGTCTTGTAAACCACTTCATTGC-3′) and reverse (5′-AGTCCGTTTAGCACGCTAC-3′) primers and 75 nM of the probe (Fam 5′-AAGAAAGATCTTTCAGGG-3′ BHQ1; underlined bases are “locked” nucleic acids). The PjHSP70a real-time PCR assay was performed using the Rotorgene 3000 (Corbett Research, Sydney, Australia) with an initial denaturation of 95°C for 8 min followed by 45 cycles of 10 s: 95°C, 20 s: 72°C and 30 s: 72°C. The results were determined by comparing unknowns with a 10-fold dilution series from ∼106 to ∼1 copy/reaction. Dilution series were made from preparations of pGEM-T easy (Promega, Southampton, UK) plasmid containing the amplicon of interest. The plasmids were linearised prior to repurification using a PCR cleanup kit (Qiagen) and quantified using Pico green reagent (Molecular Probes, Invitrogen, Paisley, UK). Reactions were set up using the CAS liquid-handling robot (Corbett Research) placed in a laminar flow hood to reduce contamination. All clinical samples and standards were run as duplicate PCR reactions.
Negative controls (ultrapure RNAse-free water, Sigma) were distributed to provide maximum information as to the potential source of any contamination. Three negative controls were aliquoted prior to dilution series, three further controls were established before aliquoting clinical samples and an additional four controls were prepared at the end. In summary, 10 negative controls were included in each PCR reaction and a total of 110 negative control PjHSP70a reactions were performed in the assessments.
Reactions were also initially assessed by agarose gel electrophoresis (3% agarose gel with TBE and ethidium bromide) and products were sized using the HYPER ladder IV and V (Bioline, London, UK). The amounts of extracted DNA used for the PjHSP70a assay were 0.5% of extract obtained by DNAeasy and 2% of extract obtained by QIAamp UltraSens. As real-time PCR results are quantitative, a receiver-operator curve (ROC) was generated from the data. No universal reference material was available with which to compare findings, so ROC analysis was used to establish the optimum cut-off which enabled discrimination between PCP and alternative diagnoses.
mt LSU rRNA
The mt LSU rRNA assay was a 25 μl reaction using 1 U Hot Taq (BioGene) with supplied buffer, 3 mM MgCl2, 200 μM each dNTP, 250 μg/μl tRNA (Sigma) and 200 nM of primers pAZ102-H and pAZ102-E (5). The mt LSU rRNA PCR assay was performed using the Palm-Cycler (Corbett Research) with an initial denaturation of 95°C for 8 min followed by 40 cycles of 30 s: 95°C, 30 s: 62°C and 60 s: 72°C. Reactions were assessed by agarose gel electrophoresis (1% agarose gel with TBE and ethidium bromide) and products sized using the 3 μl (8 ng/μl) HYPER ladder IV (Bioline).
Samples with PCR product showing band density >24 ng/400 base pairs HYPER ladder IV band were scored as positive; samples with no product or product showing the same of less density were scored as negative. Real-time PjHSP70a and conventional mt LSU rRNA PCR data were compared.
Statistical analysis
For patients with PCP, duration of anti-Pneumocystis treatment before BAL, Pao2 levels and quantity of P jirovecii DNA were log10 transformed to fit a normal distribution. Univariate analyses were performed using Spearman’s rank test for continuous variables and the Mann-Whitney test for binary variables to assess correlations between variables and the amount of detectable P jirovecii DNA in BAL fluid samples. STATA Version 9.0 was used for statistical analysis; p values <0.05 were regarded as statistically significant.
RESULTS
Molecular assay detection sensitivities
The PjHSP70a assay reproducibly detected as few as ∼5 copies/reaction (table 2) and had a dynamic range across nine orders of magnitude (fig 2A). When the same dilution experiment was performed with the mt LSU rRNA assay, the detection sensitivity was ∼105 copies/reaction despite extensive optimisation (fig 2B).

Patients
Among those with PCP, mortality at 1 month was 14.5% (9/62); all patients with alternative diagnoses survived. Among patients with PCP, Pao2 ranged from 5.1 to 13.2 kPa (median 9.6). Ten patients were receiving prophylaxis. Seven patients had pulmonary co-pathology: pulmonary Kaposi sarcoma (n = 3), C neoformans pneumonia (n = 2), pulmonary Strongyloides stercoralis infection (n = 1) and pulmonary tuberculosis (n = 1).
Detection of P jirovecii DNA by real-time PCR using the PjHSP70a assay
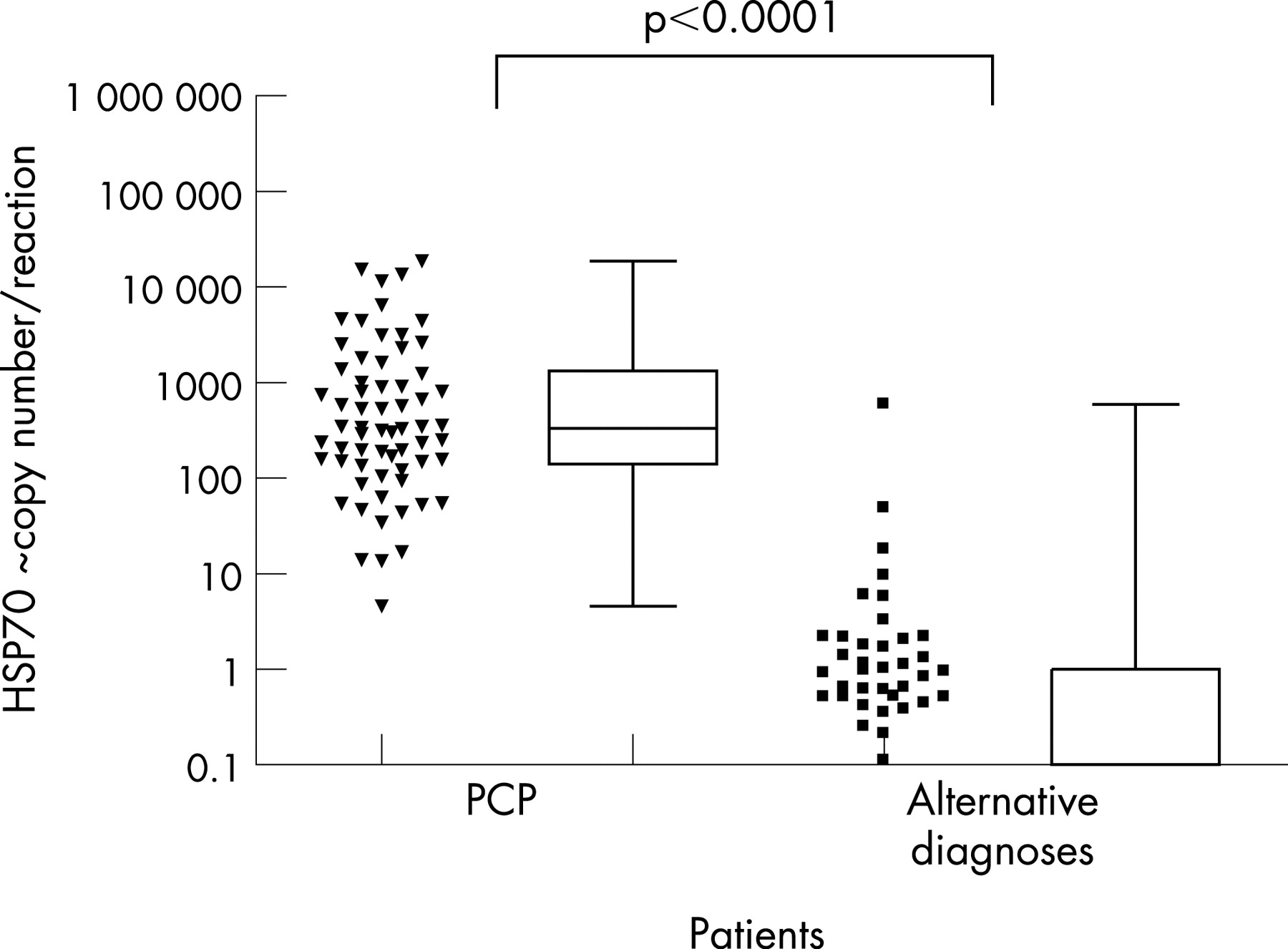
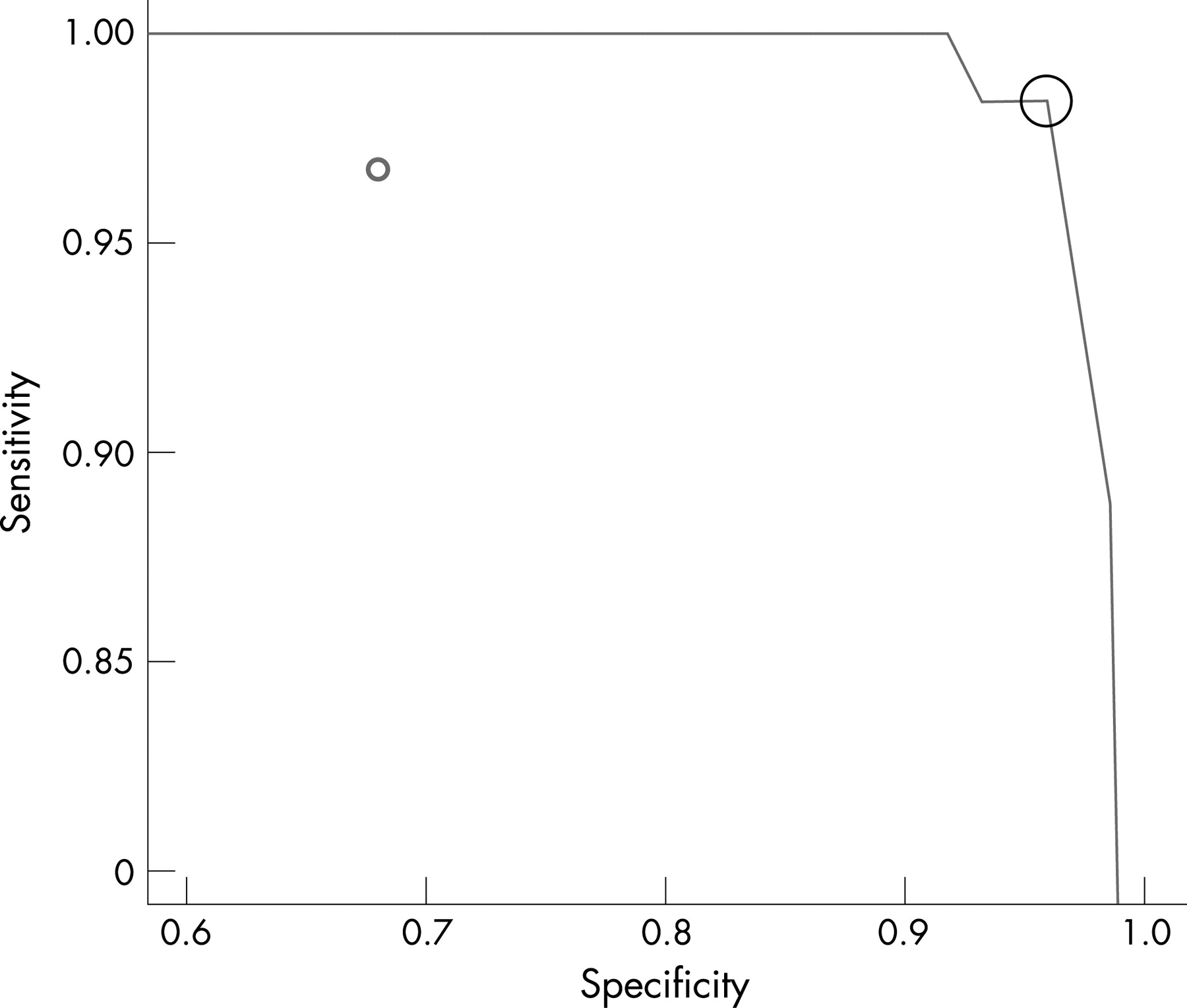
In the QIAamp-extracted samples, quantifiable P jirovecii DNA was detectable in 61/62 episodes of PCP (fig 3), ranging from ∼13 to 18608 copies/reaction (median ∼332). ROC analysis demonstrated a clinical sensitivity of 98% (95% confidence Interval (CI) 91% to 100%) and specificity of 96% (95% CI 87% to 99%) when a quantitative threshold of >10 copies/reaction was used to define a diagnosis of PCP (fig 4; sensitivity and specificity were derived from analysis of patients and not total episodes).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
One patient with PCP had detectable P jirovecii DNA but below the limits of quantification (<5 copies/reaction). This patient had a first episode PCP, a CD4 count of 0 cells/μl, had not received anti-Pneumocystis prophylaxis and had received 5 days of high-dose co-trimoxazole before BAL was performed. Necropsy confirmed PCP.
Among 71 patients (74 episodes) with an alternative diagnosis, quantifiable (>5 copies/reaction) P jirovecii DNA was detectable in 6 (8%) episodes (∼6–590 copies/reaction; median 14; table 3), detectable but below the limits of quantification in a further 34 (46%) and was undetectable in 35 (46%). Levels of detectable quantifiable P jirovecii DNA in those with PCP were higher than in those with alternative diagnoses (p<0.001, fig 3).
Among patients with PCP there was no correlation between the quantity of detectable P jirovecii DNA and patient’s prior receipt of prophylaxis, duration of anti-Pneumocystis treatment before diagnosis, Pao2 or the presence of pulmonary co-pathology. Of 110 negative controls, only one had detectable DNA (<5 copies/reaction). None of nine other negative controls in that reaction produced a detectable signal.
Detection of P jirovecii DNA using PCR at the mt LSU rRNA
Pneumocystis jirovecii DNA was detected using the mt LSU rRNA assay in BAL fluid from 61of 62 (96%) episodes of PCP and from 24 of 74 (32%) episodes with an alternative diagnosis (fig 4). Using the defined cut-off of >24 ng/reaction gel gave a clinical sensitivity of 97% (95% CI 89% to 99%) and a specificity of 68% (95% CI 56% to 78%) for a diagnosis of PCP.
DISCUSSION
This study describes the design, optimisation and performance of a real-time PCR assay intended for detection of P jirovecii. Numerous reports have described molecular assays using conventional PCR to diagnose PCP, the most common being variations on the method targeting mt LSU rRNA.2 3 10 More recently, real-time PCR has been used for the detection of P jirovecii DNA in respiratory samples and for the diagnosis of PCP.13–21 29 The present study differs from previous reports by the strategy used for optimising DNA extraction and by the gene target used for analysis. We chose an “in silico” strategy to find an optimal molecular target in the HSP70 gene and compared this method with a well described assay targeting mt LSU rRNA.2 3 10
There are many reasons for selecting this new target: the HSP70 gene is conserved across eukaryotic organisms,22 30 yet the sequence identity is not. Phylogenetic analysis suggests this sequence is unlikely to be lost, reducing the likelihood of sensitivity being compromised. As this sequence is diverse, the potential for a molecular method detecting other organisms that may be present within the sample—either as a co-pathogen or as a commensal—are greatly reduced. This PCR assay is specific for P jirovecii as it does not detect other species of Pneumocystis, including P carinii, P murina or P wakefieldiae, or a wide range of fungal, bacterial or mycobacterial potential pathogens. Furthermore, by designing the HSP70 assay to detect part of the coding sequence, polymorphisms affecting the resultant heat shock protein are also less likely than if the assay amplified a less conserved non-coding region or a highly variable protein such as the major surface glycoprotein.
When the PjHSP70a assay is used to amplify DNA from BAL fluid, the amounts of P jirovecii HSP70 DNA detected from patients with PCP and those with alternative diagnoses were significantly different. Using a quantitative cut-off of >10 copies/reaction, ROC analysis showed clinical sensitivity of 98% and specificity of 96%. Furthermore, in those with PCP, the amount of P jirovecii DNA in BAL fluid was not associated with disease severity and outcome, suggesting that other factors are implicated in the pathogenesis of PCP.
Interpretation of the significance of finding P jirovecii DNA in a respiratory sample is hampered by the observation that the organism may be asymptomatically present in the lung, either transiently or latently.7–10 31 Furthermore, a “negative” result from attempts at DNA detection in the context of asymptomatic carriage becomes more difficult to interpret when using conventional PCR. A faint band following gel electrophoresis might be due to a low starting copy number, which may or may not be attributable to “colonisation”; however, inhibition and/or low assay efficiency may also contribute to such a finding. There are further difficulties if a mitochondrial sequence such as mt LSU rRNA is used, as little is known about the physiology of P jirovecii and how the mitochondrial DNA copy number (a factor of mitochondrial numeracy and genome replication) may vary according to different states of disease, “colonisation” or in response to treatment.
We found that the mt LSU rRNA PCR assay had an analytical sensitivity for detecting P jirovecii DNA of ∼105 copies/reaction. This result is surprising and may in part be explained by the secondary structure found in both primers pAZ102-H and pAZ102-E when analysed using mfold,32 and by the 5°C difference in melting temperature between these primers identified using the nearest neighbour prediction method.33 Both of these factors could affect PCR efficiency at this locus. This finding suggests that mitochondrial sequences can be in great abundance (at least 1000 copies to every one of HSP70) when P jirovecii is present in BAL fluid, as analytical sensitivity was comparable to the single copy PjHSP70a assay. The mt LSU rRNA PCR assay can be nested to improve sensitivity, with a reported detection sensitivity of a single-copy target.34 Our data suggest that nested PCR at the mt LSU rRNA locus is unnecessary as, with an optimised extraction procedure, single round PCR at this locus has a sensitivity of 97%. Whichever conventional PCR strategy is chosen, improved analytical sensitivity will be achieved at the cost of reduced specificity, as increasing numbers of patients with low burden asymptomatic “colonisation” are detected. A quantitative method is therefore essential.
Studies which attempt to quantify P jirovecii DNA from BAL fluid and which aim to correlate genome copy numbers with clinical situations (colonisation or PCP) may potentially be confounded by the influence of variable dilution of BAL fluid caused by variations in bronchoscopic technique. In this study we attempted to control for this as one person performed all the BAL using a standard technique. Future studies which control for heterogeneity caused by dilution, either by inclusion of internal amplification controls35 or by normalising to genomic DNA,36 may enable better clinical correlations to be made.
Several real-time PCR assays using a variety of gene targets have been described for detecting P jirovecii in respiratory samples;12 16 19 a high inter-laboratory agreement among real-time PCR assays has been described.29 Alvarez-Martinez et al compared nested PCR with real-time PCR using the dihydropteroate synthase gene target applied to BAL fluid samples.19 Nested PCR had an analytical sensitivity of 94% and a specificity of 81% for detection of P jirovecii. Real-time PCR detected P jirovecii DNA from 67/70 patients with microscopically confirmed PCP, range 1–106 copies/μl of sample (mean 1.8×104) and from 3/70 with negative microscopy for PCP, range 1–10 copies/μl of sample (mean 4), giving a clinical sensitivity of 94% and a specificity of 96%.19 Larsen et al12 described development of a quantitative touch-down real-time PCR using the multi-copy major surface glycoprotein (MSG) gene for diagnosis of PCP. Lower respiratory tract samples (BAL fluid and induced sputum) from patients with PCP contained 2.4–1 040 000 copies of MSG gene/reaction (median 417) and samples from those without PCP contained 0.3–248 copies/reaction (median 2.6). Discrimination between infection and colonisation was possible by using an arbitrary cut-off value of 10 copies/reaction. However, applying this cut-off meant that the false positive detection rate fell at the cost of an increase in false negative results.12 In a follow-up study the authors applied the MSG real-time PCR assay to oral wash samples for diagnosis of PCP and demonstrated a clinical sensitivity of 88% and specificity of 85%.16 A marked treatment effect was observed in those with PCP. Samples obtained ⩽1 day after the start of treatment contained a median of 417 MSG gene copies/reaction (range 0–21 290), and samples from those who had received >1 day of treatment contained a median of 7 copies/reaction (range 0–3673). Clinical specificity was increased to 100% but sensitivity was reduced to 75% by applying a post hoc cut-off value of 50 copies/reaction.16 This observation contrasts with the findings of our study in which we found no correlation between duration of treatment prior to obtaining the BAL fluid sample and HSP70 copy number in those with confirmed PCP. In the present study, if all patients with detectable P jirovecii DNA using the PjHSP70a assay are considered positive, then clinical sensitivity would be 100% and specificity would be 37%. In order to discriminate between those who did and did not have PCP, we used a cut-off value of ∼10 copies/reaction based on ROC analysis.
Assessments of new molecular diagnostic methods are often reported on a “yes the pathogen is present” versus “no it is not present” basis, depending on whether or not a nucleic acid target is detected in a clinical sample. From such analysis, positive and negative predictive values are derived from what is often a small and highly polarised group of individuals. Diagnostic assays are often not fully characterised, making a negative result difficult to interpret as it is uncertain whether this is a true negative result or is due to sampling procedure, extraction technique or the PCR assay itself. In this study we have demonstrated the importance of DNA extraction techniques, as we compared two procedures and showed improved detection of P jirovecii DNA using QIAamp UltraSens. A large number of negative controls were used to exclude potential contamination. Only 1 of 110 negative controls had a detectable signal; this result would have been considered negative using ROC analysis. We interpret this as representing sporadic contamination (∼1% of assays).
In summary, molecular detection tests require considerable optimisation in order to be diagnostically useful. In particular, variations in DNA extraction methods may influence the amount of detectable P jirovecii DNA in BAL fluid. Real-time PCR using the HSP70 genomic target enables discrimination of PCP from other infections that form part of the differential diagnosis in immunosuppressed patients. Real-time PCR targeting HSP70 has a potential diagnostic application. A larger prospective translational study is required to define the suitability of this diagnostic approach in a routine clinical setting. The finding of P jirovecii DNA in BAL fluid in some patients without confirmed PCP is consistent with the concept of “colonisation” and underscores the need for a quantitative approach to detection.
Acknowledgments
The authors thank Professor Melanie Cushion, Veterans Affairs Medical Center, Cincinnati, USA for the gift of P carinii, P wakefieldiae and P murina DNA and for insightful advice; and Dr Jeremy Garson, Department of Virology, University College London for helpful discussions on the subject of real-time PCR.
REFERENCES
Footnotes
-
Competing interests: This research was supported by The Dr Hadwen Trust (JFH, TN, AZ and RFM).
-
Funding: Professor R F Miller is Co-Editor of Sexually Transmitted Infections, part of the BMJ Publishing Group.
Linked Articles
- Correction