Article Text

Abstract
Background: Clinical studies have shown that injurious mechanical ventilation is associated with increased airspace and plasma levels of interleukin-1β (IL-1β); however, the potential therapeutic value of IL-1 inhibition in acute lung injury has not been thoroughly investigated. A study was undertaken to determine if IL-1 signalling is a necessary early event in the pathogenesis of experimental ventilator-induced lung injury.
Methods: Mice deficient in IL-1 receptor type 1 (IL1R1) and rats treated with IL-1 receptor antagonist (IL-1Ra) were mechanically ventilated with high tidal volume (30 ml/kg) and the effect of IL-1 signalling blockade on the severity of lung injury was determined.
Results: Permeability, as measured by radiolabelled albumin flux, was significantly lower in IL1R1 null mice than in wild-type mice during injurious ventilation (p<0.05). IL-1Ra significantly decreased protein permeability and pulmonary oedema in rats during injurious ventilation and also decreased airspace and plasma levels of the chemokine CXCL1 and airspace neutrophils. IL-1Ra decreased expression of NOS2 and ICAM-1 mRNA in whole lung. Bronchoalveolar lavage fluid levels of RTI40, a marker of type I cell injury, were 2.5 times lower following IL-1Ra treatment (p<0.05). In isolated type II pneumocytes, IL-1β reduced electrical resistance and increased transepithelial permeability.
Conclusions: IL-1 contributes to alveolar barrier dysfunction in VILI by promoting lung neutrophil recruitment and by increasing epithelial injury and permeability. Because preserved alveolar barrier function is associated with better outcomes in patients with acute lung injury, these data support further testing of IL-1Ra for the treatment of acute lung injury.
Statistics from Altmetric.com
Acute lung injury, including the acute respiratory distress syndrome (ARDS), is a common cause of respiratory failure associated with 40% mortality.1 Mechanical ventilation for patients with acute lung injury is life saving, but excessive tidal volume contributes to lung injury and is associated with higher mortality. Tidal volume reduction to 6 ml/kg predicted body weight reduces mortality,2 but it is not certain if a truly safe strategy of mechanical ventilation exists. Furthermore, there are no validated diagnostic tools to aid in the recognition of ongoing ventilator-attributable lung injury. Although the mechanisms of the protective effects of tidal volume reduction are not completely known, previous clinical studies have demonstrated associations between airspace and plasma levels of biological markers of the inflammatory response and outcomes in patients with mechanically ventilated acute lung injury (reviewed by Frank et al3). For example, higher tidal volume and lower positive end-expiratory pressure (PEEP) ventilation results in persistently high plasma levels of interleukin-1β (IL-1β) and is associated with non-pulmonary organ failure and mortality.4 5 Other studies have shown that plasma levels of IL-1-inducible mediators such as IL-6, IL-8, IL-104 6 and nitric oxide synthase (NOS) activity7–9 are higher in patients ventilated with larger tidal volumes. IL-1 has been identified as the major contributor to the proinflammatory activity of bronchoalveolar lavage (BAL) fluid obtained from patients with ARDS.10 In gene expression microarray studies, IL-1β has been identified as a candidate gene, potentially important in the pathogenesis of ventilator-associated lung injury.11 Although early increases in expression levels of anti-inflammatory cytokines, soluble receptors and receptor antagonists have been reported in patients with ARDS,12 the potential therapeutic role of exogenous IL-1 receptor antagonist (IL-1Ra), a naturally occurring inhibitor of IL-1 signalling, in patients with acute lung injury has not been thoroughly investigated.
The objective of this study was to test the hypothesis that IL-1 signalling is central to the initial pathogenesis of ventilator-induced lung injury (VILI). Using IL-1 receptor type 1 null mice (IL1R1KO), we tested the effect of blocking IL-1 signalling on lung injury severity in a mouse model of VILI. We then confirmed and extended the mouse studies by treating rats with an infusion of IL-1Ra to inhibit IL-1 signalling during mechanical ventilation and measured the effect on alveolar barrier function, lung neutrophil recruitment, lung injury severity and IL-1-dependent mediator expression.
METHODS
Detailed methods are available in the online supplement.
Ventilator-induced lung injury (VILI) model
Mice and rats were anaesthetised and placed on a mechanical ventilator with a tidal volume of 30 ml/kg and without PEEP. A second group of mice was ventilated with low tidal volume (6 ml/kg) and 3 cm H2O PEEP. Animals not exposed to mechanical ventilation were used as an additional control group. Ventilation was continued for up to 3 h in the mouse studies. The rat studies were continued for an additional hour (4 h). For the rat studies, animals ventilated with high tidal volume received rhIL-1Ra or an equal volume of saline through a catheter in the internal jugular vein. A loading dose of 10 mg/kg in 0.5 ml was given before starting high tidal volume ventilation and then 10 mg/kg/h was given as a continuous infusion throughout the protocol. This dose was selected to provide a minimum of a 100-fold molar excess of IL-1Ra relative to IL-1 levels to ensure inhibition of IL-1 signalling.13
Lung endothelial and epithelial permeability to albumin and pulmonary oedema
Extravasation of intravascular 125I-labelled albumin into the extravascular spaces of the lung is reported as the radioactivity of the blood-free lung expressed as a percentage of the whole body plasma volume and normalised to non-ventilated controls. Pulmonary oedema (excess lung water) was measured using gravimetric methods.
Histological evaluation
5 μm sections were prepared and stained with haematoxylin and eosin and scored 0–4 for oedema severity, septal thickening and inflammatory cell infiltration in a blinded fashion.
IL-1β, CXCL1, nitrite and RTI40 measurements
In the mouse and rat studies, IL-1β protein levels in BAL fluid were measured using species-specific ELISAs. BAL fluid levels of RTI40 were measured in the rat samples using an immunoblot assay. In addition, BAL fluid levels of nitrite were measured in the rat samples as an index of NOS2 activity.
NOS2 and ICAM-1 gene expression
For the rat studies, mRNA expression levels of the IL-1-inducible genes NOS2 and ICAM-1 were measured in whole lung samples using semi-quantitative real-time PCR. Expression levels were quantified by comparing relative expression to GAPDH mRNA expression and in reference to internal standard curves.
Primary alveolar epithelial cell isolation and permeability assay
Rat alveolar type II cells were isolated and cultured on transwells. On day 5, serum-free medium was added to both the apical and basal compartments. The apical compartment also contained 10 μg/ml of 4 kD FITC-dextran. Recombinant rat IL-1β was then added to both sides of the monolayers. At 24 h, transepithelial electrical resistance (TEER) was measured and fluorescence was measured in a sample of medium from the basal compartment.
Statistical analysis
Comparisons among groups were made by ANOVA with the Student-Newman-Keuls post-hoc test for multiple comparisons. Comparisons within groups were made with paired t tests. Parametric comparisons were appropriate for all measures based on the Kolmogorov-Smirnov goodness-of-fit test; p values <0.05 were considered statistically significant.
RESULTS
Transgenic mouse studies
BAL fluid IL-1β protein levels in mouse VILI
BAL fluid levels of IL-1β protein were significantly increased in mice ventilated with high tidal volume for 3 h (mean (SD) 36 (32–40) pg/ml, n = 7) compared with unventilated controls (7 (6–8) pg/ml, n = 4, p<0.001). After 1 h of high tidal volume ventilation, BAL fluid IL-1 β levels were not changed (6 (5–7) pg/ml, n = 4). Low tidal volume ventilation for 3 h did not affect BAL fluid levels of IL-1β (6 (5–7) pg/ml, n = 4).
Lung albumin permeability and pulmonary oedema
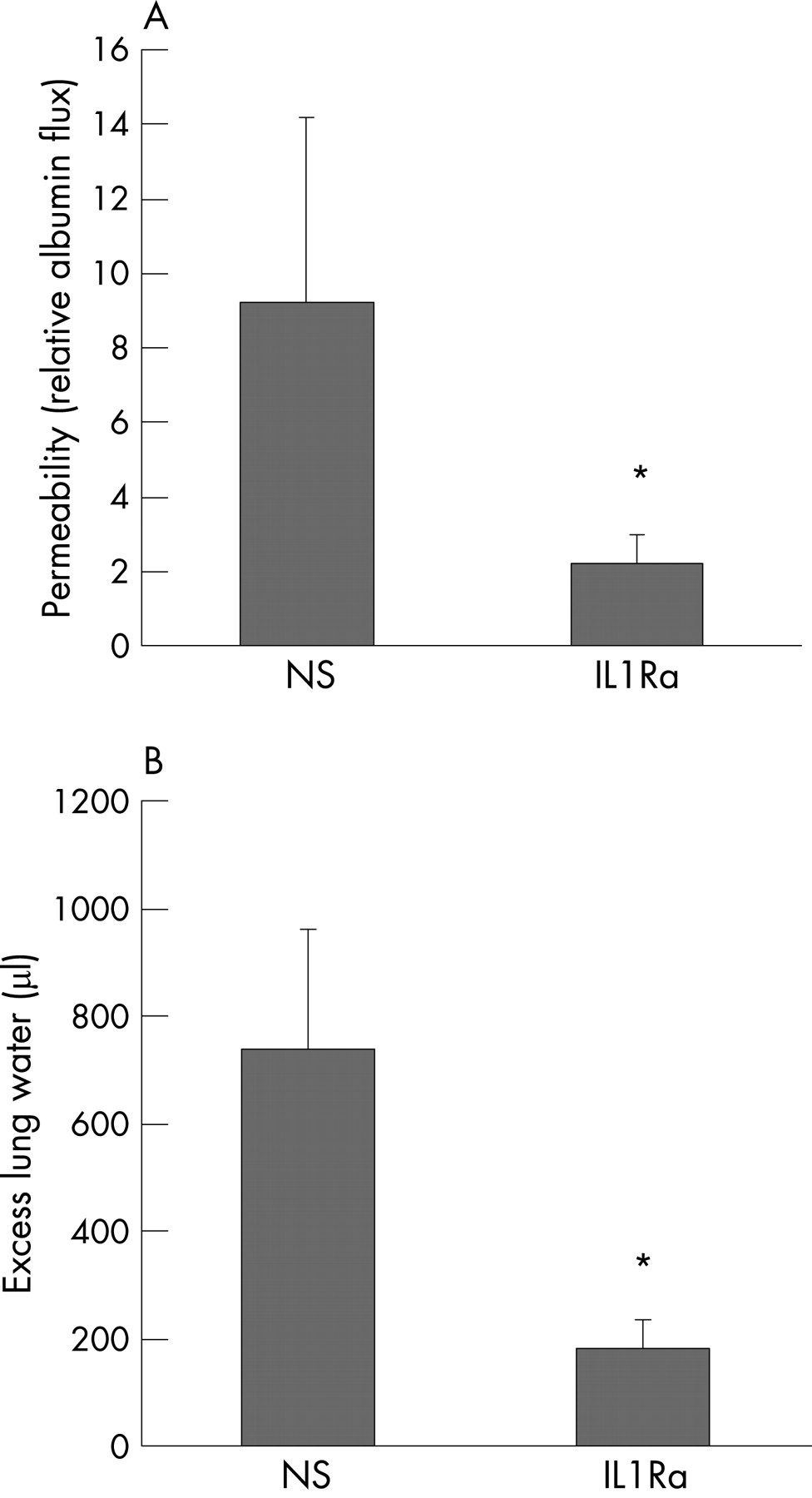
IL1R1KO mice accumulated less extravascular radiolabelled albumin following injurious ventilation than wild-type mice (n = 10 in each group; p<0.05, fig 1A). There was no difference in baseline permeability expressed as a percentage of plasma volume between uninjured IL1R1KO mice and wild-type mice (n = 10 in each group). Low tidal volume ventilation resulted in smaller matched increases in albumin permeability in both groups (p<0.05 vs unventilated controls). Similarly, there was less pulmonary oedema (excess lung water) in IL1R1KO mice following injurious ventilation (fig 1B). Peak airway pressures in IL1R1 null mice were significantly lower compared with wild-type mice following injurious ventilation while baseline respiratory mechanics were not different (not shown).

Histology and BAL neutrophil counts
Figure 1S (in the online supplement) shows representative images from each group demonstrating more severe lung injury in subjects in which IL-1 signalling was intact. Ten high-power fields from four individuals from each treatment group were scored 0–4 for oedema severity, alveolar septal thickening and inflammatory cell infiltration. The three scores for each field were added (maximum = 12) and the mean score for each individual was determined. The average scores for each group were compared by unpaired t test. The mean (SE) lung injury score for wild-type mice was 3.7 (3.2–4.2) compared with 2.0 (1.8–2.2) for IL1R1 knock out mice (p<0.05). Low tidal volume ventilation did not result in lung injury as measured by histology (not shown). Total BAL neutrophil counts were significantly lower in IL1R1KO mice. After injurious mechanical ventilation there were 1227 (370–2084) neutrophils/ml in BAL fluid from wild-type mice and 61 (13–108) neutrophils/ml in IL1R1KO mice (mean (SD), p<0.05, n = 5 in each group).
Rat IL-1Ra studies
BAL fluid IL-1β protein levels in rat VILI
As in the mouse studies, BAL IL-1β protein levels were also increased with injurious ventilation in rats compared with unventilated controls (fig 2S in online supplement). Similarly, IL-1β levels in lung homogenate were significantly increased with high tidal volume ventilation (p<0.05, n = 6 in each group). IL-1β levels were also measured in BAL fluid from IL-1Ra-treated rats. IL-1Ra treatment appeared to attenuate the increase in BAL fluid levels of IL-1β (142 (52–232), n = 5; p<0.05 vs BAL fluid levels in untreated ventilated rats).

Effect of blocking IL-1 signalling on lung albumin permeability and pulmonary oedema
Consistent with the IL1R1KO mouse data, rats treated with IL-1Ra (n = 10) had lower lung albumin permeability in the VILI model than saline-treated controls (n = 11, fig 2A). IL-1Ra-treated rats had significantly less pulmonary oedema (fig 2B).
Histology
Treatment with IL-1Ra significantly decreased the severity of lung injury (fig 3). The mean (SE) lung injury score for saline-treated rats was 6.9 (6.1–7.7) compared with 3.1 (2.9–3.3) for IL-1Ra-treated rats (p<0.05).

Effect of IL-1Ra on respiratory system compliance and oxygenation in VILI
After 4 h of injurious ventilation, dynamic respiratory system compliance decreased only in saline-treated rats (table 1). Arterial oxygen tensions decreased only in the saline-treated group. There were no differences in arterial blood pressure between the two groups of rats during the ventilation protocol (not shown).
Effect of IL-1Ra on neutrophil chemokine levels and airspace neutrophils
BAL fluid levels of CXCL1 (GRO-α/KC) were lower in IL-1Ra-treated rats than in saline-treated rats (fig 4A). Rats treated with IL-1Ra had fewer neutrophils in the BAL fluid than saline-treated controls following ventilation (fig 4B). Although total BAL fluid cell counts were similar in each group (mean (SD) 465 (380–546) ×103 cells/ml in IL-1Ra-treated rats (n = 5) and 491 (368–614] ×103 cells/ml in saline-treated rats (n = 4)), there were significantly fewer neutrophils and more macrophages in the IL-1Ra-treated group (fig 4B). The total BAL cell count in unventilated rats (n = 4) was 561 (514–608) ×103 cells/ml with 99% macrophages.

Effect of IL-1Ra on expression of IL-1-induced biological mediators
To further determine if IL-1Ra blocked IL-1 signalling, mRNA expression levels of ICAM-1 and NOS2 were measured and BAL fluid nitrite was measured as a surrogate for NOS2 activity. Following injurious ventilation, whole lung mRNA expression of ICAM-1 and NOS2 were significantly lower in rats treated with IL-1Ra compared with saline controls (fig 3S in online supplement). Mean (SD) BAL fluid nitrite levels were lower in rats given IL-1Ra (4.8 (3.3–6.3) μM vs 8.2 (6.2–10.2) μM, p<0.05; n = 5 in the IL-1Ra group and n = 4 in the saline group). BAL fluid nitrite levels in unventilated rats were 3.0 (2.2–3.8) μM.
Effect of IL-1Ra on alveolar epithelial type I cell injury in VILI
In rats, BAL fluid levels of the type I cell-specific protein RTI40 correlate with ultrastructural, histological and physiological measures of acute lung injury.14 Rats treated with IL-1Ra had lower BAL fluid levels of RTI40 after injurious ventilation than saline controls (fig 4S in online supplement).
Effect of IL-1β on transepithelial electrical resistance and permeability to 4 kD dextran in primary rat alveolar epithelial cells
In primary rat alveolar epithelial cell monolayers, IL-1β decreased transepithelial electrical resistance (fig 5A) and increased permeability to 4 kD FITC-labelled dextran (fig 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
Previous clinical studies have suggested that IL-1β may be a biological marker of VILI in patients with ARDS.4–6 10 We found that airspace IL-1β protein levels were increased early in the course of experimental VILI. The primary objective of this study was therefore to determine if IL-1 signalling is critical to the initial pathogenesis of VILI. Although increased airspace levels of IL-1β are not specific to VILI, these data show that IL-1 is an important mediator of lung injury resulting from high tidal volume, low PEEP ventilation.
The type I IL-1 receptor (IL1R1) is required for IL-1 signalling while the type 2 receptor serves a decoy function.15 Mice deficient in IL1R1 developed significantly less lung injury than wild-type mice. Alveolar barrier permeability to albumin was significantly lower in IL1R1 null mice, and these mice had less pulmonary oedema following injurious ventilation (fig 1). Histological evaluation also demonstrated less severe lung injury in IL1R1 null mice (fig 1S in online supplement). Previous experimental studies have shown that IL-1Ra may limit cytokine release and lung neutrophil recruitment in other models,16–19 raising the possibility that exogenous IL-1Ra may limit the severity of alveolar barrier disruption in VILI. To extend the mouse studies, we therefore tested the effect of pharmacological doses of IL-1Ra delivered as a continuous infusion beginning with the initiation of mechanical ventilation on lung injury severity in a rat model of VILI. Although in vivo studies in transgenic mice are valuable, there are some advantages to rat studies including improved ability to monitor circulatory physiology and arterial blood gases and more reliable administration of medications as a continuous infusion. We extended the duration of the ventilation protocol by an additional hour in the rat studies to examine the effect of IL-1Ra over a longer time period. Rats treated with IL-1Ra developed less pulmonary oedema and had lower lung permeability to albumin than saline-treated rats. In addition, arterial oxygenation was preserved and respiratory system compliance did not decrease in the IL-1Ra-treated rats, consistent with less severe lung injury (table 1).
IL-1 induces expression of IL-8, ICAM-1 and NOS2 via activation of NFκB20–22 to promote neutrophil recruitment from the circulation during injury. Previous studies have shown that VILI is dependent on the recruitment of neutrophils into the lung.23–27 We measured expression levels of CXCL1 (GRO-α, an IL-8 orthologue), ICAM-1 and NOS2 expression and activity to provide additional evidence that IL-1Ra blocked IL-1 signalling and to determine the mechanisms for the IL-1Ra-mediated protection from VILI. We found that protein levels of the neutrophil chemokine CXCL1 were lower in the airspaces of ventilated rats treated with IL-1Ra. Lung ICAM-1 and NOS2 mRNA expression levels were also lower in rats treated with IL-1Ra, and airspace nitrite levels (a marker of NOS2 activity) were lower with IL-1Ra treatment. Interestingly, one recent study found that mice deficient in NOS2 were protected from lung injury in a VILI model.28 Others have reported that NOS2 activity results in increased neutrophil sequestration in the lung in experimental sepsis.29 Taken together, these data show that IL-1 is a proximal mediator of inflammation and neutrophil recruitment in the initial pathogenesis of VILI.
In the present study, IL-1Ra treatment was associated with significantly less alveolar epithelial cell injury in the rat model. Rats treated with IL-1Ra had lower BAL fluid levels of RTI40, a type I alveolar epithelial cell-specific membrane protein. Previous studies from our group and others have shown that RTI40 levels correlate well with histological, ultrastructural and physiological measures of acute lung injury.14 Inhibition of IL-1 signalling therefore resulted in decreased lung neutrophil recruitment, decreased lung protein permeability and decreased alveolar epithelial cell injury in experimental VILI.
It is not certain if the increased IL-1β protein levels measured in the present study were the result of increased IL-1 secretion from alveolar epithelial cells or from other cell types. However, alveolar macrophages may be one important source of IL-1β protein in VILI. Alveolar macrophages are known to be a major source of IL-1 in the lungs of patients with ARDS.30 We have previously found that alveolar macrophages are activated within minutes of initiating high tidal volume, zero PEEP ventilation, and macrophage depletion results in decreased neutrophil recruitment and less severe lung injury in the rat VILI model.31 In the present study there were fewer macrophages in the BAL fluid from saline-treated rats than from IL-1Ra-treated rats (fig 4), consistent with increased activation-associated macrophage adhesion to the alveolar epithelium in the saline group. Inhibition of IL-1 signalling may therefore limit autocrine or paracrine activation of alveolar macrophages in this model.
To further explore the mechanisms for the IL-1-mediated loss of alveolar barrier function, we studied the effect of IL-1 on epithelial barrier properties in vitro. Primary rat alveolar epithelial cells were cultured on membrane supports and TEER and permeability to 4 kD FITC-dextran were measured in the presence or absence of IL-1β. Under serum-free conditions, IL-1β induced a dose-dependent decrease in TEER with a concomitant increase in permeability to labelled dextran. These data suggest that IL-1 may also act through neutrophil-independent mechanisms to disrupt alveolar barrier function in VILI, including directly increasing alveolar epithelial permeability. The molecular mechanisms of the observed increase in epithelial permeability in vitro remain to be investigated; however, previous studies have reported that IL-1 increases oxidative stress in alveolar epithelial cells32 and activates platelet activating factor—a potent mediator of pulmonary oedema.33 Interestingly, IL-1β has also been shown to decrease expression of the alpha subunit of the epithelial sodium channel (ENaC), resulting in decreased epithelial sodium transport.34 In VILI, this would be expected to decrease oedema fluid clearance from the airspaces resulting in more severe pulmonary oedema.
The processes regulating acute inflammation and the innate immune response in the lung in response to injury are complex, involving a variety of cell types and responses to several inputs such as growth factors, cytokines, chemokines, toll-like receptors and other pathways.35–40 It is not yet clear to what extent tumour necrosis factor-α and other mediators of lung injury function in parallel or in series with IL-β in this model, but blockade of IL-1 signalling significantly reduced lung neutrophil recruitment and protected the alveolar epithelium during injurious ventilation.
This study has potential implications for clinical VILI. There are currently no effective pharmacological treatments for acute lung injury. A previous phase III trial of IL-1Ra in patients with sepsis tested the effect of IL-1Ra (100 mg loading dose and up to 2 mg/h for 3 days) on 28-day mortality.41 There was no overall mortality benefit for patients receiving IL-1Ra in the entire group; however, in the subgroup of 223 patients with ARDS and sepsis, there was a significant reduction in mortality in those who received IL-1Ra (34% in patients receiving the 2 mg/h dose vs 49% in the placebo group, p = 0.04). For patients with ARDS with an expected mortality of ⩾24% (n = 150), the apparent benefit of IL-1Ra treatment on mortality was even greater (34% vs 60%, p = 0.01).41 These data were not confirmed in a subsequent study;42 however, in the second study of patients with sepsis, the subgroup of patients with ARDS was not further stratified by disease severity or predicted mortality. It is possible that the lack of a clear beneficial effect of IL-1Ra in patients with ARDS may have resulted from heterogeneity within this subgroup with respect to disease severity, as well as variability in the timing of onset of ARDS and mechanical ventilation and the initiation of IL-1Ra. Of course, neither study was designed to examine the effect of IL-1Ra on lung injury outcomes. In the case of clinical ventilator-associated lung injury, it is possible that a treatment strategy could be started before the injurious stimulus—that is, just before mechanical ventilation is started. The data from the present study are consistent with the hypothesis that early activation of the inflammatory response, including increased IL-1 signalling, is a major mechanism of alveolar barrier dysfunction in VILI. Because alveolar epithelial injury and the loss of alveolar barrier function are associated with mortality in patients with acute lung injury,43 44 this study supports further testing of the therapeutic value of IL-1Ra in these patients.
Acknowledgments
The authors thank Dr Leland Dobbs for the generous gift of the rat RTI40 antibody and Dr Jorge Gutierrez for measuring the BAL RTI40 levels.
REFERENCES
Footnotes
-
Funding: This work was supported by National Institutes of Health, National Heart, Lung and Blood Institute grants HL69900 (JAF) and HL51854 (MAM).
-
Competing interests: None.
Linked Articles
- Airwaves