Article Text

Abstract
Chronic obstructive pulmonary disease (COPD) and obesity are common and disabling chronic health conditions with increasing prevalence worldwide. A relationship between COPD and obesity is increasingly recognised, although the nature of this association remains unknown. This review focuses on the epidemiology of obesity in COPD and the impact of excessive fat mass on lung function, exercise capacity and prognosis. The evidence for altered adipose tissue functions in obesity—including reduced lipid storage capacity, altered expression and secretion of inflammatory factors, adipose tissue hypoxia and macrophage infiltration in adipose tissue—is also reviewed. The interrelationship between these factors and their contribution to the development of insulin resistance in obesity is considered. It is proposed that, in patients with COPD, reduced oxidative capacity and systemic hypoxia may amplify these disturbances, not only in obese patients but also in subjects with hidden loss of fat-free mass. The potential interaction between abnormal adipose tissue function, systemic inflammation and COPD may provide more insight into the pathogenesis and reversibility of systemic pathology in this disease.
Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) and obesity are major causes of morbidity and mortality worldwide and, according to current estimates, the global burden of these conditions will increase further. While spirometry is used for diagnosis and gradation of COPD severity according to the Global initiative for chronic Obstructive Lung Disease (GOLD) guidelines,1 classification of overweight and obesity is based on body mass index (BMI) as defined by the World Health Organization (WHO).2
According to the Burden of Obstructive Lung Disease (BOLD) Initiative,3 the estimated international prevalence of stage II or higher COPD is currently 10.1% in individuals aged 40 years and older. COPD was the fifth leading cause of mortality around the world in 2001 and will be the third most frequent cause of death by the year 2020.4 The prevalence of obesity, defined as BMI ⩾30 kg/m2,2 has multiplied during the last decades and varies from 10–20% in most European countries to >32% in the USA.2 According to the most recent global WHO projections, 400 million adults were obese in 2005 and it is estimated that this number will exceed 700 million by the year 2015.5 Obesity plays a major role in the development of the metabolic syndrome and has been identified as an important risk factor for chronic diseases such as type 2 diabetes mellitus and cardiovascular disease. It is linked to respiratory diseases such as obstructive sleep apnoea syndrome and obesity hypoventilation syndrome,6 and accumulating evidence suggests an association between obesity and asthma.7 A potential link between obesity and COPD is also increasingly recognised,8 although very little is known about the mechanisms underlying this association. The risk of developing obesity is increased in patients with COPD as a result of a reduced level of physical activities in daily life in these patients compared with healthy age-matched controls.9 In addition, in theory, patients with COPD who receive repeated courses of systemic glucocorticosteroids are at increased risk of truncal obesity as a result of glucocorticoid-mediated redistribution of stored energy and the stimulatory effect on intake.10
PREVALENCE OF OBESITY IN COPD
Steuten et al11 studied the prevalence of obesity in a large primary care population of patients with COPD in the Netherlands. The overall prevalence of obesity in this population was 18%, with the highest prevalence in GOLD stages 1 and 2 (16–24%) and the lowest in GOLD stage 4 (6%). For comparison, the current prevalence of obesity in the general population in the Netherlands is 10% in adult men and 12% in adult women.12 A much higher prevalence of obesity was reported by Eisner et al13 in an adult multi-ethnic cohort of patients with early stage COPD in Northern California, USA; 54% of the patients with COPD had a BMI ⩾30 kg/m2, which is considerably more than the 20–24% of obese individuals in the same US state reported by the Centers for Disease Control and Prevention using annual data from the Behavioural Risk Factor Surveillance System (www.cdc.gov/BRFSS/). Thus, available data suggest that obesity is more prevalent in patients with COPD than in the general population, depending on the severity of chronic airflow limitation. Indeed, a study in moderately severe patients participating in cardiopulmonary rehabilitation showed that abdominal obesity measured by waist circumference was almost twice as common in patients with COPD as in age- and sex-matched controls.14 In addition, the occurrence of obesity might differ between various clinical phenotypes of COPD. The classical distinction of “pink puffers” and “blue bloaters” was based on anthropometric characteristics of patients with emphysema and chronic bronchitis, respectively. By definition, “pink puffers” were thin in appearance with a frequent history of weight loss while “blue bloaters” showed no marked weight loss and tended to be overweight.15 In fact, a significantly increased prevalence of (pre)obesity (BMI ⩾28 kg/m2) was reported in patients with chronic bronchitis (25%) compared with controls (16%) in the Tucson prospective cohort study,16 while underweight was more prevalent in patients with emphysema.
There are no published studies on the prevalence of COPD in obesity. However, data from the 4th examination of The Copenhagen City Heart Study suggest that the prevalence of COPD in obese individuals is significantly lower than in the rest of the population (11.2% vs 18.6%) owing to a lower occurrence of mild and moderate disease (personal communication, Jørgen Vestbo, Copenhagen, Denmark).
In addition to obesity characterised by an absolute abundance of fat mass in patients with COPD, it is well recognised that alterations in body composition also commonly occur in normal weight patients with COPD. Selective wasting of fat-free mass (FFM; ie, muscle mass) occurs in 10–15% of normal weight patients with COPD,17 18 resulting in a relative or absolute increase in fat mass.19 This situation shows a resemblance to the involuntary loss of muscle mass with advanced age defined as sarcopenia.20
Box 1 Respiratory function in eucapnic obesity
Decreased chest wall and lung compliance
Small airway dysfunction and expiratory flow limitation
Preservation of the forced expiratory volume in 1s/forced vital capacity ratio
Variable reduction of (or preserved) ventilatory muscle strength and endurance
Increased work and oxygen cost of breathing
Normal or increased carbon monoxide transfer factor
Abnormal ventilation/perfusion relations and arterial oxygen desaturation
Normal or increased central respiratory drive
EFFECT OF OBESITY ON RESPIRATORY FUNCTION AND EXERCISE PERFORMANCE
In any given individual the effects of obesity on respiratory function depend on the mass and anatomical distribution of the excessive adipose tissue on the thorax and abdomen.21 22 The nature of the physiological impairment induced by obesity will vary with its extent and will ultimately reflect integrated abnormalities of ventilatory mechanics/muscle function, pulmonary gas exchange, ventilatory control and cardiac performance.21–35 These respiratory derangements are consistently accentuated on adopting the supine posture. Common physiological abnormalities associated with eucapnic obesity (excluding obesity hypoventilation syndrome) are listed in box 1. Restrictive mechanics are manifest by reduced expiratory reserve volume and functional residual capacity. Residual volume and total lung capacity are preserved except in extreme obesity. The forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) ratio is preserved but tests of small airway function may be abnormal.21–35 Lung carbon monoxide transfer factor (Tlco) is in the normal range or increased and gas exchange is compromised in severe obesity, particularly in the supine posture. Normative predictive equations for spirometric measurements, plethysmographic lung volumes and Tlco are generally not corrected for weight. This has implications for the clinical interpretation of pulmonary function tests in the overweight and obese who comprise an increasing proportion of those tested.
Exercise performance
Peak oxygen uptake (V.o2) corrected for weight is surprisingly preserved in obesity, at least during weight-supported cycle exercise.27 36–38 When comparing oxygen uptake measurements during exercise in obese individuals with predictive normal values, underestimation in the former can be avoided by expressing V.o2 as a percentage of ideal body weight. Obese individuals have a higher metabolic demand at any given power output as a result of the high oxygen cost of lifting heavy limbs.27 36–38 Pulmonary gas exchange is rarely critically compromised in obesity and arterial oxygen saturation may even improve during exercise, reflecting an improvement in ventilation/perfusion relationships.27 36–38 Patients with obesity may have expiratory flow limitation at rest which, when compounded by high ventilatory requirements, leads to significant air trapping and dynamic increase in end-expiratory lung volume during exercise (fig 1).27 37 38 Given the increase in ventilatory demand (secondary to increased metabolic loading) and the excessive intrinsic mechanical loading of the inspiratory muscles in obesity, the muscular effort required to support a given ventilation is increased at any given power output compared with individuals of normal weight.38–40 Not surprisingly, the work and oxygen cost of breathing is also increased at rest and during exercise.36 39 41 42 Exertional dyspnoea intensity is uniformly increased for any given power output and rises sharply as ventilation (and therefore contractile muscle effort) approach maximal capacity.27 In some patients, exercise performance is further compromised by attendant skeletal muscle deconditioning, peripheral muscle dysfunction and cardiac impairment (eg, eccentric left ventricular hypertrophy of obesity).43 44
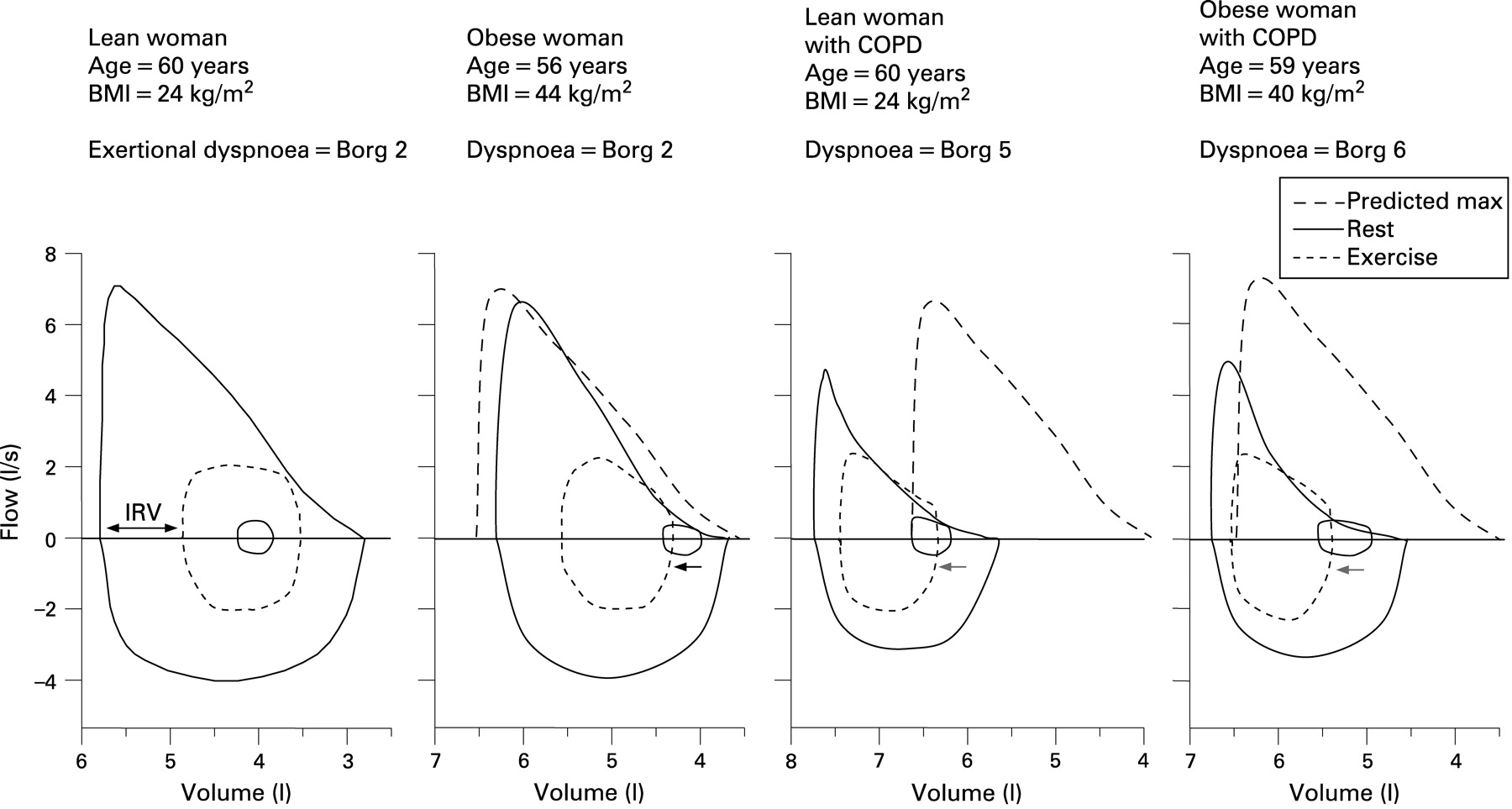
Combined deleterious effects of obesity and COPD
The physiological derangements that occur at rest and during exercise in obesity and in COPD are well understood when each is considered in isolation. However, no mechanistic studies have explored the pathophysiological interactions that occur when both conditions coexist in the same individual, as is increasingly the case. Given the vast pathophysiological heterogeneity of COPD, the concomitant effects of aging and sex on physical performance, and the existence in many of serious comorbidities in conjunction with obesity, the mechanisms of exercise intolerance in obese patients with COPD are necessarily complex. Clinical experience indicates that the combined restrictive/obstructive deficits evident in obese patients with COPD culminate in worsening symptomatology and activity limitation.8 45 46 Obesity would be expected to amplify the abnormalities of dynamic ventilatory mechanics and ventilatory demand that characterise COPD. Thus, ventilatory requirements are predictably higher in obese patients with COPD compared with those of normal weight as a result of the increase in chemostimulation that arises from the combination of increased metabolic loading, high fixed physiological dead space and possibly earlier metabolic acidosis due to impairment in oxygen uptake from deconditioning or cardiac dysfunction. The increased recoil of the chest wall and lung in patients with central obesity may increase expiratory flow rates at a given operating lung volume compared with non-obese patients with COPD. However, the relatively reduced absolute end-expiratory lung volume in obese patients with COPD may worsen expiratory flow limitation and air trapping during exercise (fig 1), particularly in the setting of the relatively increased ventilatory requirement.47 48 The net effect of these abnormalities is an earlier mechanical limitation of ventilation with greater dyspnoea and exercise curtailment in the obese patient with COPD. The accompanying rapid shallow breathing pattern (at a relatively low power output and ventilation) may compromise dynamic inspiratory muscle function and deleteriously affect alveolar ventilation.47–49 In obese patients with COPD the increased contractile muscle effort required to sustain ventilation during exercise in the face of serious mechanical constraints on tidal volume expansion would be expected to cause significant neuromechanical uncoupling of the respiratory system and lead to incapacitating dyspnoea earlier in exercise than in non-obese patients with COPD.
Effects of body composition on exercise performance in obese COPD
In addition to ventilatory limitations, alterations in body composition contribute to exercise intolerance in patients with COPD. It is well recognised that loss of FFM contributes to muscle weakness50 and reduced exercise capacity51 in patients with moderate to severe COPD. Available data in these patients suggest that the contribution of alterations in fat mass to exercise intolerance is limited compared with the impact of muscle wasting. The degree of physical impairment, assessed by the 12 minute walking test, was greater in normal weight patients with depleted FFM and relatively enhanced fat mass than in underweight patients with preserved FFM.17 Less is known about effects of obesity or relatively increased fat mass on physical performance in patients with less severe COPD. In patients with early stage COPD, obesity, decreased lean-to-fat mass ratio and increased sagittal abdominal diameter were associated with functional limitation on the six minute walk test.13 The accumulation of fat mass and not the loss of lean mass had a particularly negative impact on performance. This observation is not unique for patients with early stage COPD. In a cohort of elderly individuals from the general population, high body fatness was associated with increased risk of self-reported mobility-related disability, while low FFM was not predictive of disability.52 In another community-based cohort study of older individuals, higher fat mass was related to an increased risk of functional limitation assessed by walking speed.53 Although absolute lean mass was not related to physical performance, a higher ratio of lean mass to fat mass was predictive of walking speed. Thus, it seems that both absolute and relative accumulation of fat mass contribute to functional limitation in the elderly and early in the course of COPD, while the influence of muscle wasting on physical performance becomes apparent in patients with more severe COPD when FFM falls to very low levels.17 54
THE OBESITY PARADOX IN COPD
The prognostic value of nutritional status is well established in patients with COPD. Low BMI is associated with increased all-cause and COPD-related mortality, unrelated to disease severity.55 Recent studies indicate that the FFM index is an even more important determinant of prognosis in moderate to severe COPD than BMI.18 54 Fat mass index was not predictive for prognosis in patients with moderate to severe COPD.54 Remarkably, the relative risk for mortality seems decreased in overweight and obese patients with COPD in GOLD stage 3–4, while it is increased in those with GOLD stage 1–2 disease with (pre)obesity.55 A possibly protective role for obesity in COPD was also observed in early studies on the association between body weight and mortality,56 in the epidemiological Copenhagen City Heart Study18 and in a population of patients with severe COPD treated with long-term oxygen therapy.57 This possible association between (pre)obesity and improved outcome in COPD contrasts with epidemiological data from the general population where obesity is associated with a largely decreased life expectancy, independent of smoking status.58 This phenomenon is referred to as a “reverse epidemiology of obesity” or the “obesity paradox” and was identified in several other chronic disease states such as end-stage chronic kidney disease, chronic heart failure and rheumatoid arthritis.59 The pathophysiological mechanisms of the obesity paradox are yet to be unravelled. In obese men with COPD the annual decline in FEV1 was significantly lower than in men of normal BMI range while this effect was not seen in women.60 This suggests a gender-specific protective role for obesity in the progression of chronic airflow limitation. Furthermore, it is not yet clear whether excessive fat mass or muscle mass contributes to the survival advantage in chronic wasting diseases.59
In apparent contradiction to the possible existence of an obesity paradox in COPD, it is well recognised that the risk of cardiovascular mortality is increased in patients with chronic airflow limitation, independent of BMI.61 Epidemiological studies indicate that low FEV1 is a risk factor for coronary heart disease in subjects without pulmonary disease.62 Besides the presence of chronic airflow obstruction, low-grade systemic inflammation is one of the mechanisms that may be responsible for the increased rate of cardiovascular complications in COPD.63 The role for smoking-related systemic inflammation is evident in the pathogenesis of COPD and led to the proposal of the term “chronic systemic inflammatory syndrome” complementary to the diagnosis of COPD.64 Insulin resistance may also contribute to the increased cardiovascular mortality in patients with COPD. It plays an important role in the pathogenesis of type 2 diabetes mellitus and it clusters with a variety of risk factors for cardiovascular disease, such as abdominal obesity, dyslipidaemia and hypertension in the metabolic syndrome.65 In a recent meta-analysis of longitudinal studies, the presence of the metabolic syndrome was strongly associated with cardiovascular morbidity and mortality.66 Indeed, some studies suggest the presence of insulin resistance in COPD,67 especially in normal weight to obese patients68 69 and those who are hypoxaemic.69
Based on the evidence outlined above, it can be hypothesised that obesity exerts divergent effects in subgroups of COPD with different severity of disease. Obesity may protect against mortality in patients with advanced COPD in whom loss of FFM is a particularly important short-term risk factor for death. By contrast, in earlier stage COPD the harmful long-term effects of obesity-related conditions such as low-grade systemic inflammation and insulin resistance may result in increased cardiovascular and all-cause mortality.
The implications of obesity for the pharmacological and extrapulmonary management of patients with COPD are still unknown. In recent years, new insights into the functioning of adipose tissue in obesity have evolved. These are reviewed in the next section and may help to further unravel the clinical significance and pathophysiology of obesity in COPD.
ADIPOSE TISSUE DYSFUNCTION IN OBESITY AND THE INFLUENCE OF HYPOXIA
The association between abdominal obesity, insulin resistance and cardiovascular disease is well recognised,70 but the underlying mechanisms are not yet fully understood. Adiposity is associated with insulin resistance even over relatively normal ranges of body fatness. There is strong evidence that altered adipose tissue function plays a crucial role in the pathogenesis of obesity-related insulin resistance and type 2 diabetes, as has recently been reviewed.71
Lipid storage capacity of adipose tissue in obesity
Adipose tissue is the main lipid storage depot in the body and is of crucial importance in buffering the daily influx of dietary fat entering the circulation. One characteristic of adipose tissue in obesity is the enlargement of adipocytes which may represent impaired adipocyte differentiation. The hyperthrophic adipocytes of obese subjects are overloaded with stored triacylglycerol and it is likely that the buffering capacity for further lipid storage in these adipocytes is decreased, especially in the postprandial state.72 Non-adipose tissues are therefore exposed to an excessive influx of lipids which could lead to ectopic fat deposition when the oxidative capacity is insufficient. It is well established that accumulation of lipids in skeletal muscle,73 pancreatic islets74 and the liver75 play an important role in the development of insulin resistance and/or impaired insulin secretion in obese individuals. Another possible explanation for the observation that adipocyte hypertrophy is associated with insulin resistance and the development of type 2 diabetes relates to the endocrine function of adipose tissue.
Adipose tissue as a source of inflammation in obesity
Adipocytes express and secrete a variety of adipokines—including cytokines, growth factors, adiponectin, resistin, adipsin, leptin, acylation stimulating protein (ASP), plasminogen activator inhibitor-1 (PAI-1), lipoprotein lipase (LPL) and components of the renin-angiotensin system76—which may exert local and systemic effects. Chronic low-grade inflammation is a hallmark of obesity, insulin resistance and type 2 diabetes.77 78 Importantly, inflammatory pathways play a prominent role in the mechanisms underlying insulin resistance and type 2 diabetes in cultured cells and animal models,79–81 and this may also be the case in humans.
The inflammatory response that is often present in obesity appears to be triggered and to reside predominantly in the enlarged adipose tissue.82–85 The expression and/or secretion of inflammatory molecules, including tumour necrosis factor α (TNFα),84 86 87 interleukin (IL)6,86–88 PAI-189 and leptin,87 90 are increased in the adipose tissue of obese insulin-resistant individuals. In contrast, the expression of the insulin-sensitising factor adiponectin is reduced in obese and insulin-resistant subjects.91 92 Interestingly, enlarged adipocytes seem to express pro-inflammatory and anti-inflammatory factors with a shift towards dominance of pro-inflammatory adipokines compared with smaller adipocytes.93
Recent data indicate that adipose tissue of obese individuals is infiltrated by macrophages, which may be a major source of locally produced pro-inflammatory adipokines.94 95 There seems to be cross-talk between adipocytes and macrophages within adipose tissue. For example, macrophage-secreted factors exert effects on adipocytes that may increase systemic inflammation and insulin resistance associated with obesity.96 The mechanisms underlying the impaired adipokine secretion and macrophage infiltration in adipose tissue in obesity remain to be elucidated, but there is evidence to support a major role for adipose tissue hypoxia in these events.
Adipose tissue hypoxia in obesity
Evidence has recently emerged that adipose tissue hypoxia is present in obese mice.97 Accordingly, it has been shown that weight loss decreases adipose tissue expression of hypoxia-responsive genes.98 An alteration in oxygen pressure and/or oxygen content of the blood was not apparent in the obese animals.97 Based on the above, it can be speculated that hypoxia in adipose tissue may contribute to obesity-related insulin resistance.
Since adipose tissue blood flow per unit tissue mass is reduced in obese humans99 100 and rodents,97 decreased blood supply to adipose tissue may explain adipose tissue hypoxia in obesity. Indeed, it has been suggested that the expansion of adipose tissue mass during the progressive development of obesity may lead to hypoxia in certain parts of adipose tissue because angiogenesis is insufficient to maintain normoxia in the entire adipose tissue depot.101 As a consequence, adipose tissue hypoxia may induce increased secretion of inflammatory factors, acute phase proteins and angiogenic factors by adipose tissue in order to increase blood flow and vascularisation. The transcription factor hypoxia inducible factor-1 (HIF-1) is a key regulator in the response to alterations in oxygen tension and modulates the expression of genes that are involved in angiogenesis, erythropoiesis, inflammation and glucose metabolism.102 103
Recent studies have shown that adiponectin and PPARγ mRNA expression were decreased, whereas PAI-1 and visfatin mRNA expression were increased in hypoxic 3T3-L1 adipocytes compared with normoxic control cells.97 104 105 Likewise, hypoxia altered the expression and secretion of inflammation-related adipokines in human adipocytes.106 Furthermore, weight loss led to improved oxygenation and reduced inflammation in dietary obese mice.107 Second, severe hypoxia may induce adipocyte death. A hypoxic environment devoid of nutrients prevents the cell undergoing energy-dependent apoptosis and cells become necrotic.108 Subsequently, macrophages fuse and form syncytia that sequester and scavenge adipocyte debris.109 Hypoxia-induced adipocyte death may thus evoke macrophage infiltration in the adipose tissue of obese individuals. These data suggest that adipose tissue hypoxia modulates—either directly or indirectly via recruitment of macrophages—adipokine expression and secretion and may therefore provide an important link between obesity and insulin resistance.71 Third, hypoxia inhibits adipocyte differentiation110 which appears to be a precipitating factor in the development of type 2 diabetes, as mentioned earlier.111 Finally, unfolded protein response, an HIF-1-independent signalling pathway, contributes to cellular adaptation to hypoxia.112 Many disturbances including hypoxia cause accumulation of unfolded proteins in the endoplasmatic reticulum (ER)112 113 leading to ER stress.114 It has been shown that ER stress is increased in the adipose tissue and liver of obese mice, which activates the inflammatory response, thereby contributing to insulin resistance.115 In conclusion, adipose tissue hypoxia—possibly due to impaired adipose tissue blood flow as a consequence of adipose tissue expansion—may be an important factor in the development of obesity-related insulin resistance via effects on adipokine expression and/or adipocyte differentiation.71
In summary, evidence suggests that enlargement of adipocytes, impaired adipose tissue blood flow, adipose tissue hypoxia, local inflammation and macrophage infiltration in adipose tissue are interrelated in obesity and may lead to disturbances in adipokine secretion and excessive fat storage in non-adipose tissues, which together may result in insulin resistance and ultimately type 2 diabetes mellitus.71
POTENTIAL IMPACT OF ADIPOSE TISSUE DYSFUNCTION ON THE PATHOPHYSIOLOGY OF COPD
Adipose tissue and fat oxidative capacity in COPD
There is a lack of studies on adipose tissue morphology in patients with COPD. While available evidence suggests an increased prevalence of abdominal obesity at the macroscopic level,14 no microscopic data are available in COPD. As a consequence, possible alterations in size, differentiation and lipid buffering capacity of adipocytes remain unknown.
If fatty acid delivery to skeletal muscle exceeds fat oxidation, lipids and lipid intermediates accumulate in skeletal muscle cells. Intramyocellular fat accumulation is closely related to the development of insulin resistance.116 Reduced peripheral skeletal muscle fat oxidative capacity is consistently reported in patients with COPD.117 It is reflected by a muscle fibre type shift from type I to II (or slow-twitch to fast-twitch) and reduced activities of enzymes involved in oxidative energy metabolism.118 In the case of high non-esterified fatty acid (FFA) release into the circulation in COPD, the reduced fat oxidative capacity would be a potential risk factor for development of metabolic syndrome. Since obesity is associated with increased exposure of skeletal muscle to triacylglycerol and FFA, obese patients with COPD would be at particular risk. Indeed, increased fasting lipolytic rate and enhanced levels of circulatory FFA have been reported in hypoxaemic patients with COPD in comparison with controls,119 although another study did not confirm this.120 Also, reduced intramyocellular triglyceride content was reported in a small group of normal weight patients with COPD. However, levels of oxidative enzymes were not reported in that study and no obese patients with COPD were included.
Adipose tissue as a source of inflammation in COPD
In addition to pulmonary inflammation, persistent low-grade systemic inflammation is frequently demonstrated in patients with COPD. Increased levels of pro-inflammatory cells and mediators have been reported in the circulation of patients with COPD. Enhanced plasma concentrations of fibrinogen, C-reactive protein (CRP), TNFα and circulating leucocytes have been consistently observed in patients with stable COPD compared with healthy controls,121 while raised levels of IL6,68 IL8,122 IL1068 and IL18123 have also been shown. The presence of low-grade systemic inflammation was associated with several systemic consequences in patients with COPD, including reduced exercise capacity and worse health-related quality of life.124 Also, increased circulatory levels of TNFα, IL6 and their soluble receptors were observed in FFM-depleted patients with COPD in comparison with patients with preserved FFM, both in patients with low body weight and in those with normal BMI.125 This suggests a role for systemic inflammation in COPD-related muscle wasting, even in patients without apparent weight loss such as subgroups of sarcopenic or obese patients. In addition, a link between systemic inflammation and markedly increased cardiovascular mortality in COPD was suggested.63
Despite the growing awareness that systemic inflammation is a hallmark in the pathogenesis of COPD, remarkably little is known about the underlying mechanisms. Although it is often hypothesised that inflammation in the systemic compartment is the result of spill-over of the inflammatory process in the airways, lung parenchyma and pulmonary vasculature, evidence from cross-sectional studies indicates no correlation between pulmonary and circulatory inflammatory markers in stable COPD.122
Adipose tissue is another possible source of systemic inflammation. As discussed above, adipose tissue expresses several pro-inflammatory mediators with potential systemic metabolic effects. The importance of these adipokines released from fat tissue in the pathogenesis of systemic inflammation in obesity is recognised. By measurements of arteriovenous concentration differences of cytokines in combination with measurement of adipose tissue blood flow, release of IL6 from abdominal subcutaneous adipose tissue into the systemic circulation has been demonstrated in healthy human subjects.88 Furthermore, the rate of IL6 release per unit abdominal subcutaneous adipose tissue has been shown to be equal or greater in obese subjects, depending on the degree of obesity.126 Taking into consideration the absolute increased fat mass in the obese, these findings indicate that IL6 release from abdominal subcutaneous adipose tissue is raised at the whole-body level in these individuals. In another study in obese subjects, IL6 concentrations in the portal vein, which drains the visceral fat, were significantly increased compared with IL6 levels in peripheral arterial blood, demonstrating that visceral fat is an important source of IL6 in obesity.127 Furthermore, portal vein IL6 levels were related to arterial C-reactive protein concentrations, suggesting a mechanistic link between visceral fat mass and systemic inflammation in vivo.
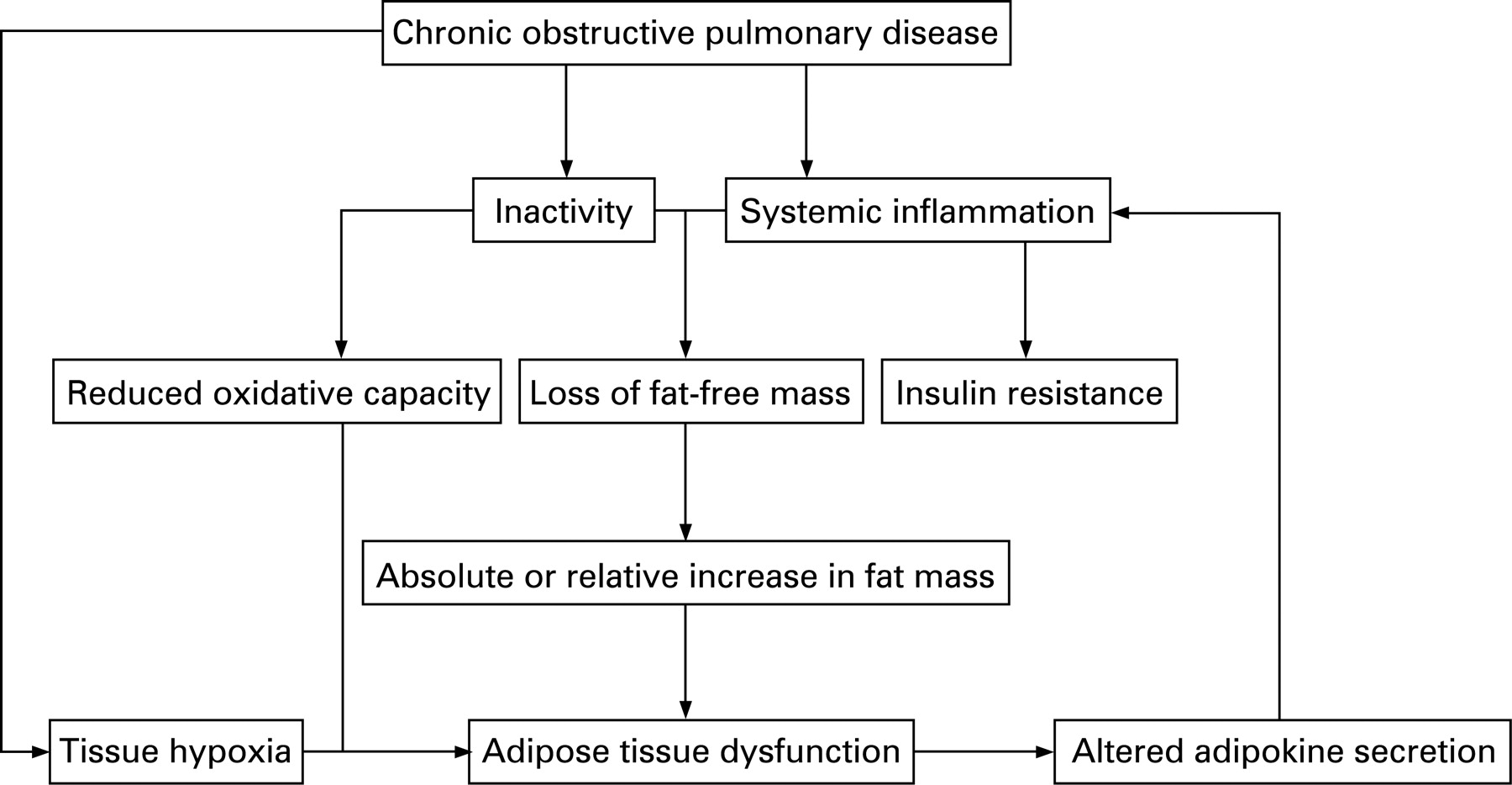
The role of adipose tissue in the pathogenesis of systemic inflammation in patients with COPD has not been studied. Also, the possible cross-talk between fat mass-derived inflammatory factors and loss of FFM in COPD is speculative. However, based on the foregoing, it can be hypothesised that, in patients with early stage COPD with obesity or relatively abundant fat mass, adipose tissue may act as an additional source of inflammation (fig 2). Since systemic inflammation is associated with loss of skeletal muscle in COPD,125 this may contribute to loss of FFM and amplify alterations in body composition towards loss of lean body mass and preservation of fat mass. Additionally, excretion of adipokines by adipose tissue in COPD might be reinforced by the presence of chronic or intermittent hypoxia.

{kind=link}
{kind=link}
Adipose tissue hypoxia and insulin resistance in COPD
As mentioned earlier, there is evidence for adipose tissue hypoxia in obesity.97 It has recently been shown that hypoxia induces expression and secretion of several pro-inflammatory mediators including IL6, leptin, PAI-1 and monocyte migration inhibitory factor (MIF) in human adipocytes.106 As stated above, adipose tissue hypoxia might thus provide an important link between obesity and insulin resistance through secretion of inflammatory adipokines.
In patients with COPD with obesity or relatively abundant fat mass, the link between adipose tissue hypoxia and systemic metabolic effects might be further enhanced by the presence of systemic hypoxaemia in more advanced stages of disease, which does not occur in healthy obese individuals. Although no studies have directly focused on this issue, there is growing evidence for insulin resistance in COPD, especially in normal to overweight and hypoxic patients. Glucose intolerance was first reported in normal weight patients with chronic hypoxaemic COPD on long-term oxygen therapy, although the results might have been confounded by the use of systemic glucocorticosteroids.128 More recently, the presence of insulin resistance was reported in normal weight patients with COPD with normal systemic oxygen saturation.67 Interestingly, both BMI and the systemic IL6 concentration were independent determinants of insulin resistance. In line with this observation, there is evidence for decreased insulin sensitivity in a large group of normal weight patients with COPD in comparison with underweight patients and normal weight controls.68 However, insulin sensitivity was not related to BMI in that study. Finally, supplemental oxygen resulted in increased glucose tolerance and insulin sensitivity in hypoxaemic patients with COPD, and this effect tended to be greater in obese than in lean subjects.69
Potential mechanistic links between adipose tissue mass, local and systemic hypoxia, systemic inflammation and insulin resistance in patients with COPD offer challenging research opportunities, not only in the extrapulmonary manifestations of the disease but also in the area of pathogenesis of obesity-related metabolic disturbances. Future studies need to reveal the potential effects of excessive fat mass on pathophysiology and prognosis in COPD and possible implications for pharmacological management, dietary interventions and pulmonary rehabilitation.
REFERENCES
Footnotes
Competing interests: None.