Article Text

Abstract
The case history is presented of a male infant who was thought to have idiopathic pulmonary arterial hypertension (PAH) at 3 months of age. Subsequently the PAH decreased unexpectedly and diffuse pulmonary arteriovenous malformations (PAVMs) were seen at 6.9 years of age for the first time. Hereditary haemorrhagic telangiectasia type 1 (HHT1) related to an endoglin mutation was diagnosed. At 10.3 years of age a lung biopsy showed diffuse PAVMs as well as pulmonary arteriopathy with medial hypertrophy. This is the first case of HHT1 presenting with PAH at such a young age. The subsequent decrease in pulmonary arterial pressure (PAP) was probably caused by the development of PAVMs. In the presence of PAVMs, measurement of the PAP may underestimate the extent of PAH-related vasculopathy.
Statistics from Altmetric.com
Pulmonary arterial hypertension (PAH) is defined as a mean pulmonary artery pressure (mPAP) of ⩾25 mm Hg at rest or ⩾30 mm Hg during exercise, with an increased pulmonary vascular resistance index (PVR) of ⩾3 Wood units (WU).m2 and a normal pulmonary artery wedge pressure (PAWP) of ⩽15 mm Hg. The currently used clinical classification of PAH (Venice, 2003) distinguishes between idiopathic sporadic and familial PAH and PAH related to cardiac or systemic disease, and a number of risk factors and associated conditions for PAH have been identified.1 In the past, the idiopathic form of PAH in childhood had a particularly poor prognosis with a median survival of <1 year.2 Mutations in genes encoding members of the transforming growth factor (TGF)-β receptor have been identified in some of these patients, indicating an aetiological role for the TGF-β pathway.3
CASE REPORT
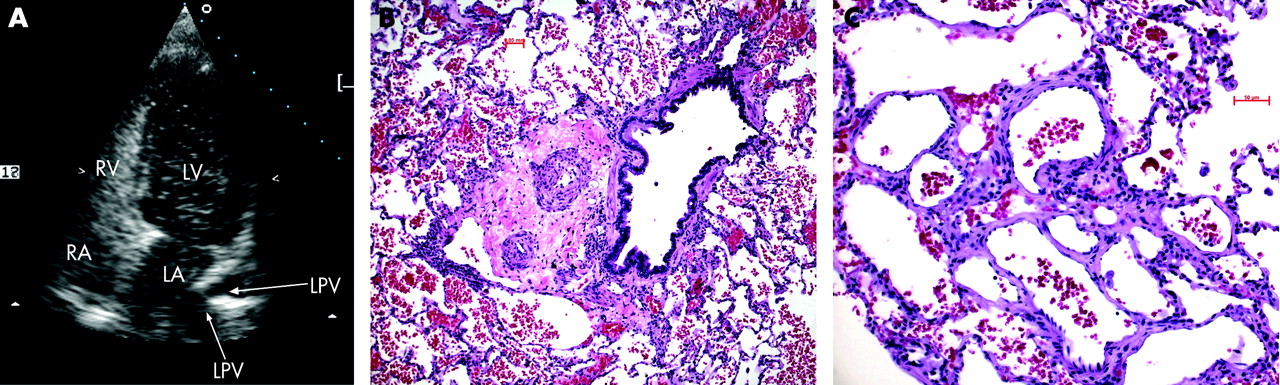
A male infant presented with recurrent episodes of cyanosis and dyspnoea since birth. At 13 weeks, cardiac catheterisation showed a raised mPAP (45 mm Hg), high PVR (15.4 WU.m2) and normal PAWP (3 mm Hg). Owing to his age and the absence of either congenital heart disease or chronic lung disease, the diagnosis of idiopathic PAH was made.1 Home oxygen therapy and oral nifedipine were administered. During follow-up, PAH-related symptoms and oxygen requirement decreased unexpectedly. At 2.9 years cardiac catheterisation showed a decrease in mPAP (25 mm Hg) and PVR (5.2 WU.m2). Doubts about the idiopathic form of PAH were raised and treatment was discontinued. At 6.9 years a decrease in exercise tolerance with progressive dyspnoea, cyanosis and headache on exertion led to cardiac re-catheterisation. The mPAP (25 mm Hg) and PVR (4.5 WU.m2) were basically unchanged. In contrast to previous pulmonary angiographic studies, on this occasion multiple pulmonary arteriovenous malformations (PAVMs) were suspected and confirmed by contrast echocardiography (fig 1). The PAVMs were below the detection limit of contrast spiral computed tomography (<3 mm). As a consequence of persistent borderline PAH, nifedipine was restarted. The gradual development of cutaneous telangiectases and occasional nose bleeds raised the suspicion of hereditary haemorrhagic telangiectasia (HHT). A branch-site mutation of endoglin (ENG IVS12-22T→C) was detected.3 Clinical deterioration led to re-evaluation at 10.3 years, demonstrating a stable mPAP (24 mm Hg) and PVR (4.3 WU.m2) with progression of PAVMs (<3 mm). For pathological assessment of PAH, a lung biopsy was performed and showed diffuse PAVMs as well as pulmonary arteriopathy with medial hypertrophy (fig 1).4 Bosentan treatment was added.5 6

{kind=link}
DISCUSSION
Familial PAH and HHT are autosomal dominant disorders of the vascular system caused by mutations of genes encoding TGF-β receptor components including bone morphogenetic protein receptor type 2 (BMPR2), activin receptor-like kinase 1 (ALK1) and endoglin (ENG).7 8 Heterozygous mutations in BMPR2 have been identified in the majority of familial PAH cases and up to 26% of patients with sporadic PAH.7 HHT1 is related to mutations in ENG.8 In HHT2, mutations in ALK1 are seen.8 The association between PAH and HHT occurs predominantly in HHT2.9 HHT1-related PAH has been reported in three adults.9 10 However, these patients had conditions or risk factors that might have contributed to the development of PAH, such as pulmonary embolism, extensive liver arteriovenous malformations with increased cardiac output and a raised PAWP, or a history of dexfenfluramine intake.9 10 In contrast, our patient with early-life PAH in HHT1 provides evidence that defective endoglin signalling by itself may lead to PAH. The parental observation of recurrent cyanosis since birth is in accordance with the hypothesis that PVR may not decrease normally after birth in some neonates with PAH being diagnosed at a later date as the pulmonary vascular disease progresses.7 In our patient PAH preceded the diagnosis of PAVMs. This clinical course raises the issue whether PAH promoted the early development of PAVMs in this patient with a genetic susceptibility.
HHT is characterised clinically by recurrent epistaxis, mucocutaneous telangiectases and visceral arteriovenous malformations. More than 20% of patients with HHT develop PAVMs, ranging from diffuse PAVMs to large complex structures.8 In our patient the decrease in mPAP was associated with the development of PAVMs. It is probable that PAVMs were present before the age of 6.9 years because microscopic PAVMs cannot be excluded by pulmonary angiography. An unexpected decrease in mPAP in a patient with PAH should therefore alert the clinician to screen for PAVMs with highly sensitive contrast echocardiography.8 Conversely, in PAVMs pulmonary haemodynamics may not reflect the extent of PAH-related vasculopathy. In PAVMs and borderline PAH, histological examination may help to assess the severity of the pulmonary arteriopathy.
Footnotes
Competing interests: None.