Article Text

Abstract
Granulomatous inflammation in lung biopsies is a relatively non-specific finding that can occur in a range of inflammatory and neoplastic conditions. This review focuses on the patterns of granulomatous inflammation that can cause diffuse lung disease, highlighting histopathological features helpful in differential diagnosis.
Statistics from Altmetric.com
CLASSIC HYPERSENSITIVITY PNEUMONIA (EXTRINSIC ALLERGIC ALVEOLITIS)
Hypersensitivity pneumonia (-itis), also called extrinsic allergic alveolitis, is a diffuse interstitial lung disorder that results from sensitisation to inhaled organic dusts or aerosols. Offending agents are typically derived from thermophilic bacteria, moulds and plant or animal proteins. Criteria for diagnosis usually include an appropriate exposure history associated with subjective and objective evidence of lung disease temporally linked to antigen exposure, and the presence of serum antibodies directed against the suspected antigen.1–3 Confirming the diagnosis is difficult, in part because the presence of precipitating antibodies reflects exposure and not necessarily disease. False negative results are also common. Given the absence of a diagnostic “gold standard”, lung biopsies often play a pivotal role in diagnosis.4
Hypersensitivity pneumonia is often divided into acute, subacute and chronic forms. The terms have been inconsistently applied, reflecting significant overlap between these interrelated categories. Acute hypersensitivity pneumonia, the term reserved for patients suffering a first attack with symptom duration <1 month, follows exposure to relatively large doses of the responsible antigen and is the most easily recognised variant. Lung biopsy is rarely necessary for diagnosis or management and, for that reason, pathological findings are incompletely described. Rare reports describe acute bronchopneumonia with an associated necrotising small vessel vasculitis.5 6 A single report illustrates a bronchiolocentric granulomatous process, but details regarding the circumstances of exposure are lacking.7
Chronic hypersensitivity pneumonia, the term applied to patients with progressive respiratory complaints for at least 1 year, results from intermittent exposure to the offending antigen and is the consequence of a type IV hypersensitivity reaction. Affected patients may be unaware of the environmental exposure responsible for their symptoms. Most patients present with the insidious onset of slowly progressive respiratory complaints, usually cough and shortness of breath.8 Conventional radiographs typically show a non-specific pattern of ground glass and reticular opacities, often with an upper lobe distribution.9 10 Volume loss and honeycomb change are present in long standing disease. High resolution computed tomography (HRCT) scans show a distinctive combination of ground glass opacities, small centrilobular nodules and mosaic attenuation.7 11–13 Irregular lines and architectural distortion in the form of traction bronchiectasis and bronchiolectasis frequently accompany the other findings. Associated honeycomb change is present in about half of patients, and may result in a pattern of infiltrates indistinguishable from usual interstitial pneumonia.13–15
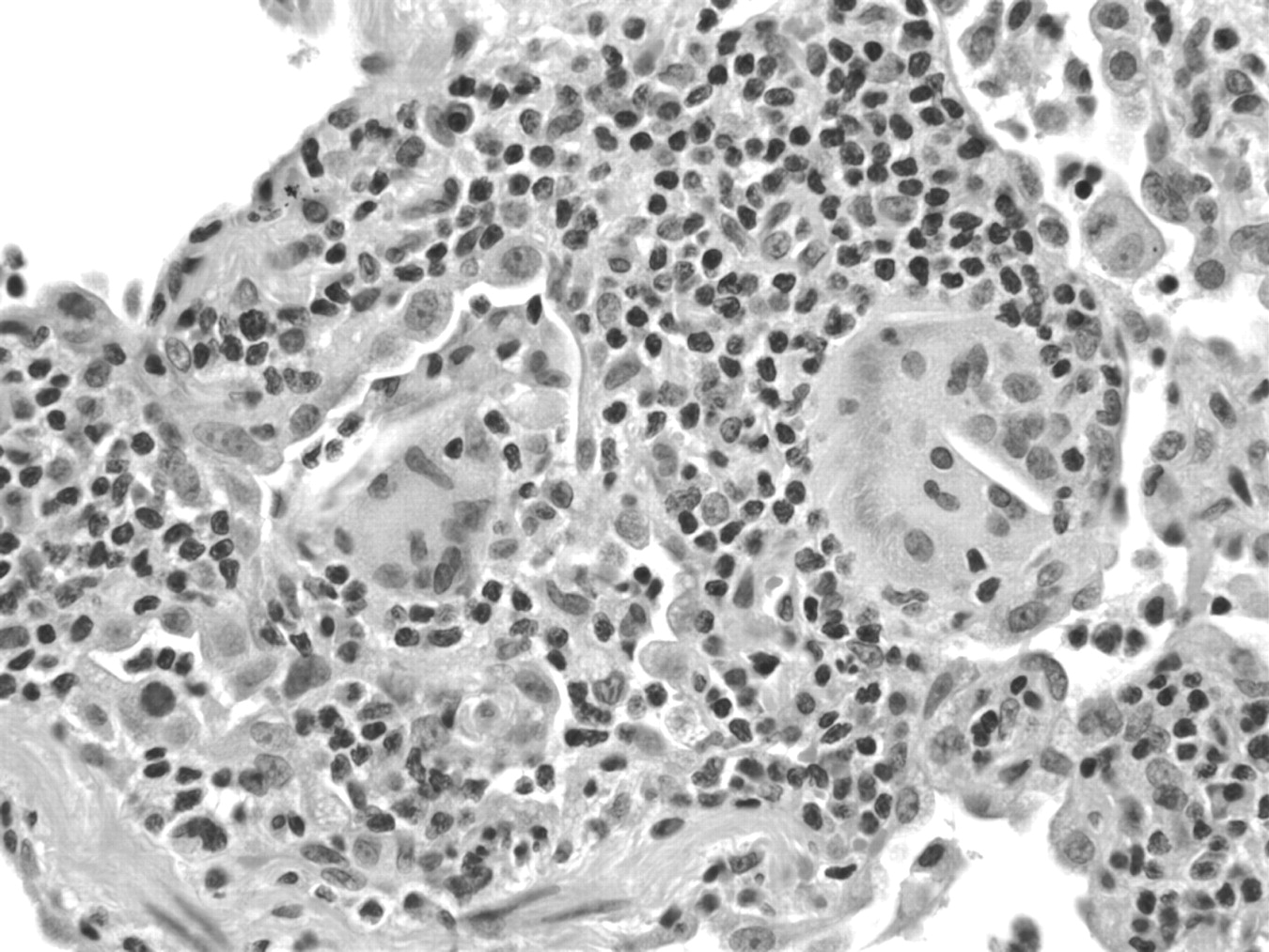
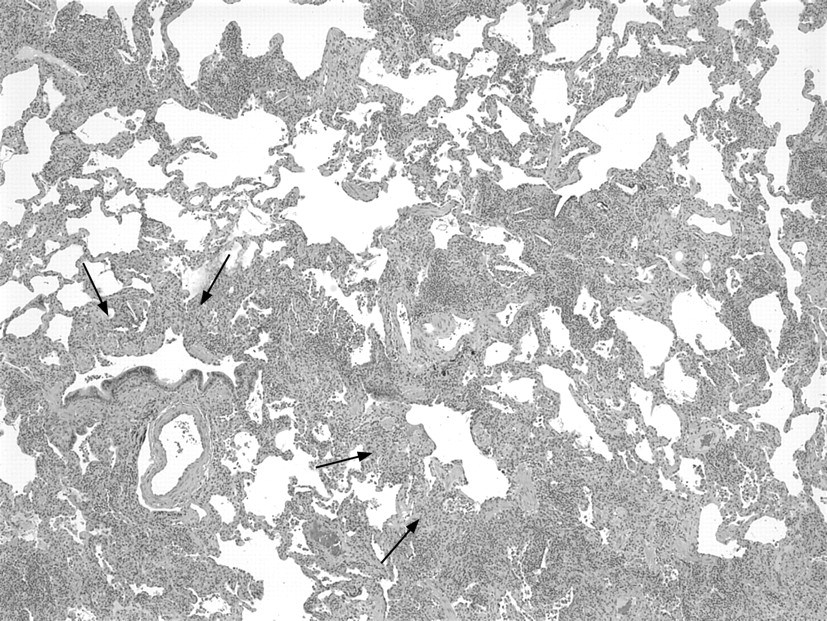
Histological diagnosis of hypersensitivity pneumonia is predicated on recognition of a combination of (1) interstitial pneumonia, (2) bronchiolitis and (3) granulomatous inflammation.16–18 Chronic interstitial pneumonia is the most consistent finding and is present in nearly all cases.3 17–26 At low magnification, alveolar septa are expanded by an infiltrate of mononuclear inflammatory cells accentuated around bronchioles (fig 1). Epithelioid histiocytes predominate only focally, usually around the airways, resulting in a subtle granulomatous appearance (fig 2). Eosinophils are usually absent or inconspicuous. Well-formed (sarcoid-like) granulomas are rare. Isolated multinucleated giant cells are common and, in a minority of cases, represent a striking feature. Giant cells often contain non-specific cytoplasmic inclusions identical to those seen in sarcoidosis such as Schaumann bodies, asteroid bodies, cholesterol clefts and birefringent calcium salts (fig 3). These inclusions are frequently misconstrued as “foreign bodies” and can give the false impression that inflammation results from inhalation or aspiration of foreign particles. Granulomatous features may be absent in as many as 30% of surgical lung biopsies.22 In rare cases, fibrosis may be severe and mimic that seen in usual interstitial pneumonia.
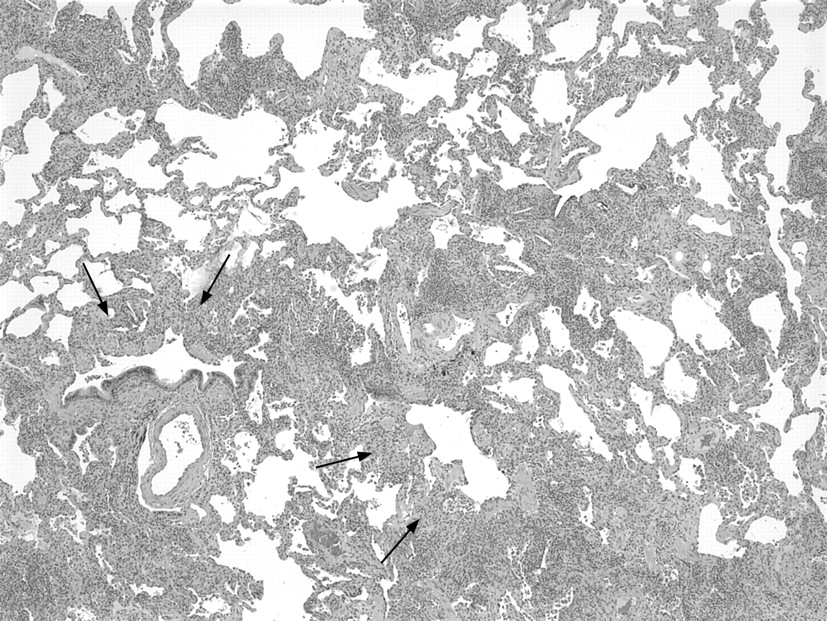

The bronchiolitis consists of a cellular peribronchiolar infiltrate of mononuclear cells, often accompanied by lymphoid aggregates.25 27 Bronchiolitis obliterans, often accompanied by organising pneumonia, is present about half the time.18 19 22 23 Small airway dysfunction results in microscopic foci of obstructive pneumonia characterised by clusters of finely vacuolated foamy alveolar macrophages within the peribronchiolar interstitium and air spaces.
DIFFUSE ATYPICAL MYCOBACTERIAL REACTIONS (“HOT TUB LUNG”)
Well-formed granulomas are rare in hypersensitivity pneumonia and, if present, should raise a suspicion of other entities. “Hot tub lung” is a syndrome that combines features of hypersensitivity pneumonia and Mycobacterium avium complex (MAC) infection resulting from exposure to contaminated indoor hot tubs, spas, jacuzzis or whirlpools.3 28–32 Affected patients complain of dyspnoea, fever and weight loss. Radiologically diffuse infiltrates are identified in 70–100% of patients. Prominent nodularity may be present. The HRCT scan shows ground glass attenuation and scattered nodules in most patients.
Lung biopsy findings resemble those seen in more classic examples of hypersensitivity pneumonia except that the interstitial pneumonia tends to be less conspicuous and the granulomas are well formed and frequently distributed within airway lumens rather than in the peribronchiolar interstitium (fig 4).30 In rare cases there may be necrosis in the granulomas. Organising pneumonia is present in about half of patients. Cultures and a history of exposure are necessary for diagnosis, as the organisms are infrequently identified on special stains of tissue sections.

SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause with worldwide distribution and protean clinical manifestations.33 Lung involvement occurs in over 90% of patients and is the dominant feature in most.33 34 Diagnosis usually requires demonstration of characteristic granulomatous inflammation in an appropriate clinical context.
Discrete well-formed non-necrotising granulomas are the histological hallmark of sarcoidosis.35–37 Granulomas comprise a compact circumscribed cluster of epithelioid and multinucleated histiocytes with minimal or no central necrosis. They are often associated with a peripheral infiltrate of lymphocytes and a distinctive pattern of lamellar fibrosis (fig 5). Giant cells frequently contain non-specific cytoplasmic inclusions such as asteroid bodies, basophilic Schaumann bodies and birefringent crystalline particles corresponding to calcium oxalates and other calcium salts.38 39 In some cases, birefringent particles may be sufficiently prominent to raise concerns for occupational lung disease. Aside from mild inflammation and variable degrees of fibrosis in the immediate vicinity of the granulomas, the remainder of the lung tissue is relatively unaffected without significant associated interstitial pneumonia. Over time, fibrosis may obliterate the granulomas and partially obscure the diagnostic features.

Distribution of the granulomas in a lymphangitic pattern is a distinctive feature of sarcoidosis that can be extremely helpful in diagnosis (fig 6). Granulomas are situated within the interstitium rather than air spaces and involve predominantly bronchovascular bundles and interlobular septa. Airway involvement is nearly universal and occasionally results in airflow limitation.40–42 Pleural involvement is less conspicuous but can be a dominant site of disease.43–48 Non-necrotising granulomatous “vasculitis” is common in surgical lung biopsies (fig 7).49–51 Vascular involvement rarely results in pulmonary hypertension.52–55 Pulmonary hypertension in sarcoidosis is more commonly a consequence of fibrosis and end stage cystic change.


Granulomas occasionally coalesce to form macroscopic nodules, a variant referred to as “nodular sarcoidosis”. Nodular sarcoidosis is characterised by large well-circumscribed nodules measuring up to 5 cm in diameter.56–58 The nodules are usually multiple and bilateral, and mimic the appearance of metastatic disease or other nodular processes such as Wegener granulomatosis.58 Central cavitation and ring-shaped opacities have been demonstrated in a few patients.59 60 Isolated solitary pulmonary nodules are rare.61 62
The main differential diagnosis in sarcoidosis is granulomatous infection, which can also occur as a complication of underlying sarcoidosis.63–66 Extensive necrosis and/or intraluminal location of granulomas are important clues to a diagnosis of granulomatous infection. Special stains and cultures should be performed in all cases of suspected sarcoidosis.
NECROTISING SARCOID GRANULOMATOSIS
Necrotising sarcoid granulomatosis (NSG) was first proposed as a unique entity by Liebow in 1973.67 It overlaps clinically and histologically with nodular sarcoidosis. Indeed, many consider NSG a subset of nodular sarcoidosis distinguished by broad zones of infarct-like parenchymal necrosis.68 69 The combination of well-formed confluent granulomas coalescing to form nodules, infarct-like parenchymal necrosis and granulomatous vasculitis are the histological hallmarks of NSG.70 Granulomatous infection is an more important consideration in NSG and should be vigorously excluded using a combination of special stains and cultures.
WEGENER GRANULOMATOSIS
Wegener granulomatosis is a vasculitic syndrome of unknown aetiology with variable clinical manifestations.71 72 The clinical spectrum ranges from mild smouldering illness to fulminant respiratory and renal failure characterised by an explosive onset and a rapidly fatal course.72–80 Classic Wegener granulomatosis involves the lungs, upper respiratory tract and kidneys. This classic combination occurred in only 14 of 50 patients reported by DeRemee and colleagues, who emphasised that each of these sites can be involved individually or in virtually any combination.81 Numerous other extrathoracic sites can also be involved. Lung involvement occurs in as many as 75% of patients over the course of their illness.77 80 81 Patients with lung disease are usually symptomatic and complain of cough, haemoptysis, dyspnoea and/or fever. “Limited Wegener granulomatosis” was defined historically as disease that involves the lungs without associated glomerulonephritis.82 More recently the term has been applied to patients who satisfy the American College of Rheumatology diagnostic criteria but without involvement that threatens the function of a vital organ or the patient’s life.83 Findings on chest imaging studies are variable but usually include multiple nodules, frequently with cavitation.84–88
Antineutrophil cytoplasmic autoantibodies (ANCA) with specificity for proteinase-3 (c-ANCA) are relatively specific for Wegener granulomatosis and represent the single most important addition to laboratory analysis of patients suspected of having Wegener granulomatosis.78 89–98 c-ANCA titres are positive in about 90% of patients with active generalised disease, in 60–85% of patients with active localised disease and in 30–35% of patients with quiescent disease.97 99–101 Serum c-ANCA titres can be used as a diagnostic tool and to monitor disease activity in patients with established diagnoses. Positive p-ANCA titres, often reflecting autoantibodies directed against myeloperoxidase, are less specific but are positive in some patients with Wegener granulomatosis.
Lung biopsy findings in Wegener granulomatosis are variable.82 102–107 Classic cases comprise a distinctive pattern of necrotising granulomatous inflammation combined with necrotising vasculitis. Broad geographical zones of necrosis are surrounded by a polymorphic inflammatory infiltrate that includes palisaded histiocytes and multinucleated giant cells (fig 8). The central necrosis has a characteristic “dirty” basophilic appearance resulting from karyorrhexis and lysis of neutrophils (“nuclear dust”). Multinucleated giant cells have a characteristic darkly staining and smudged appearance. The vasculitis involves arteries and veins of various sizes as well as capillaries (“capillaritis”). The nature of the vascular infiltrate is variable, ranging from necrotising granulomas to fibrinoid necrosis with neutrophils. The vascular changes tend to be punctate and focal (“segmental”) and can be missed with inadequate sampling (fig 9).


Some authors have focused attention on a peculiar form of necrosis (“pathergic necrosis”) as the sentinel lesion of Wegener granulomatosis.108 Proponents of this viewpoint hypothesise that necrosis of stromal collagen is followed by a neutrophilic infiltrate with formation of granulomatous microabscess. Microabscesses expand and coalesce to form broad zones of geographical necrosis. Granulomatous microabscesses are a frequent finding in Wegener granulomatosis and can be a helpful clue to the diagnosis (fig 10).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Several histopathological variants have been described, emphasising the protean manifestations of Wegener granulomatosis. The eosinophilic variant differs from more classic cases only in that the inflammatory background includes prominent numbers of eosinophils.109–111 Peripheral eosinophilia is uncommon; most patients present with clinical and radiological findings typical of Wegener granulomatosis. Airway involvement can be the predominant finding in some patients and can mimic bronchocentric granulomatosis.112 113 Pulmonary haemorrhage with capillaritis tends to be associated with a rapidly progressive course resembling Goodpasture syndrome.114–116 Katzenstein and colleagues called attention to yet another variant in which a pattern of intraluminal fibrosis resembling cryptogenic organising pneumonia (BOOP) is the dominant finding.117
OTHER FORMS OF “ANGIITIS” AND “GRANULOMATOSIS”
The now outdated term “angiitis” and “granulomatosis” was historically intended to group NSG and Wegener granulomatosis with Churg-Strauss syndrome (CSS), bronchocentric granulomatosis and lymphomatoid granulomatosis.118 CSS is unique in that lung biopsies invariably demonstrate eosinophilic pneumonia as the dominant feature while granulomas and necrotising vasculitis are inconsistent findings.119 120 Bronchocentric granulomatosis (BCG) refers to a non-specific phenomenon in that virtually any granulomatous process, including granulomatous infection and Wegener granulomatosis, can be centred on large and small airways. BCG is the term most frequently applied to a form of bronchocentric necrosis commonly seen in allergic bronchopulmonary aspergillosis (ABPA).121 Mucoid impaction of bronchi and eosinophilic pneumonia usually accompany the BCG in this setting and are important clues to the diagnosis. Lymphomatoid granulomatosis is not a truly granulomatous process but rather a form of malignant lymphoma in which a distinctive pattern of necrosis mimics the appearance of necrotising granulomatous inflammation.122
GRANULOMAS IN OTHER CONDITIONS
Granulomas occasionally occur as a non-specific or incidental finding in other unrelated conditions including various neoplasms. Granulomas resembling those seen in sarcoidosis have been observed in association with carcinomas, carcinoid tumors and various forms of malignant Hodgkin and non-Hodgkin lymphoma. In the mediastinum, seminoma (dysgerminoma) is another neoplasm in which granulomatous inflammation can be a prominent confounding feature.
SUMMARY
Granulomatous inflammation is a relatively non-specific histopathological phenomenon. Characterisation of other histological features coupled with an understanding of the clinical and pathological context are key to diagnosis of specific disease entities.
REFERENCES
Footnotes
Funding: None.
Competing interests: None.