Article Text

Abstract
Smoking-induced diffuse interstitial lung processes include respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease (RBILD), desquamative interstitial pneumonia (DIP) and Langerhans’ cell histiocytosis. The histological, radiological and clinical features of respiratory bronchiolitis, RBILD and DIP are reviewed, with particular reference to management issues; Langerhans’ cell histiocytosis is covered elsewhere in this series of articles. Possible relationships between smoking and other diffuse lung diseases are explored briefly.
- BAL, bronchoalveolar lavage
- DIP, desquamative interstitial pneumonia
- HRCT, high resolution computed tomography
- IPF, idiopathic pulmonary fibrosis
- NSIP, non-specific interstitial pneumonia
- RBILD, respiratory bronchiolitis-associated interstitial lung disease
- Tlco, carbon monoxide transfer factor
- UIP, usual interstitial pneumonia
Statistics from Altmetric.com
- BAL, bronchoalveolar lavage
- DIP, desquamative interstitial pneumonia
- HRCT, high resolution computed tomography
- IPF, idiopathic pulmonary fibrosis
- NSIP, non-specific interstitial pneumonia
- RBILD, respiratory bronchiolitis-associated interstitial lung disease
- Tlco, carbon monoxide transfer factor
- UIP, usual interstitial pneumonia
Respiratory bronchiolitis is a very common (and possibly invariable) incidental finding in cigarette smokers, consisting of an accumulation of pigmented macrophages within respiratory bronchioles and adjacent alveoli.1 This characteristic histological picture and the often associated findings of peribronchiolar inflammation and fibrosis were first identified in a necropsy study in 1974 that included 19 young asymptomatic smokers.2 It was not until 1987 that a clinically significant diffuse lung disease was described in association with respiratory bronchiolitis. Myers et al3 reported six heavy smokers with lung crackles, a restrictive ventilatory defect and chest radiographic abnormalities indicative of interstitial lung disease. Histological examination of open lung biopsy specimens disclosed clusters of macrophages in and around terminal bronchioles and thickening of the surrounding alveolar septa with a mild chronic inflammatory cell infiltrate. As these findings were confirmed,4,5 the clinical distinctions between respiratory bronchiolitis, increasingly regarded as an incidental finding in smokers, and smoking-related interstitial lung diseases were refined. The term “respiratory bronchiolitis-associated interstitial lung disease” (RBILD)5 was promulgated to distinguish between the airway centred interstitial entity first described by Myers et al3 and the more diffuse disorder, desquamative interstitial pneumonia (DIP). It was subsequently suggested by Katzenstein that RBILD should replace DIP as a unifying term, based on the perception that RBILD and DIP are overlapping manifestations of the same disease process,6 a view that is not universally shared.
More recently, as it was accepted that the spectra of histological abnormalities in respiratory bronchiolitis and RBILD are identical, RBILD came to be viewed as an exaggerated respiratory bronchiolitic response resulting in cough, shortness of breath and pulmonary function impairment.
RESPIRATORY BRONCHIOLITIS, RBILD AND DIP: RELATIONSHIP WITH SMOKING
Respiratory bronchiolitis is almost invariably associated with smoking. In a landmark histological study, Fraig et al7 evaluated 156 surgical lung biopsy specimens in which appearances were not perturbed by another underlying disease and the smoking history was considered to be reliable. Typical respiratory bronchiolitis was evident in 108 patients including, strikingly, in all 83 current smokers but in none of the 24 non-smokers. It was also present in 25 of 49 former smokers, including some who had not smoked for 5 years or longer. The level of cytoplasmic pigmentation of macrophages and the presence of peribronchial fibrosis correlated with the pack-year smoking history. In some earlier reports, current and former smokers were grouped together in analysis and therefore findings of occasional smokers without respiratory bronchiolitis are not incompatible with the findings of Fraig et al.7 In spite of this limitation, Cottin et al8 found respiratory bronchiolitis in 89% of 79 smokers with pneumothoraces. Remy-Jardin et al9 observed pigmented macrophages in 69% of 41 resected lungs in current smokers but did not view this finding as indicative of respiratory bronchiolitis; thus, differences in histological diagnostic criteria are an important confounding factor. The balance of evidence suggests that respiratory bronchiolitis represents a highly specific physiological response to smoking.
No compelling reports exist of typical respiratory bronchiolitis in the absence of smoking. In the index series of Niewohner et al,2 5 of 24 patients were categorised as non-smokers but the smoking histories in this necropsy study were necessarily taken from relatives; the absence of subsequent reports of respiratory bronchiolitis in non-smokers suggests that former smoking was not always rigorously excluded. The peribronchial pigmented macrophages accumulating in dust-related environmental lung diseases lack the characteristic brown pigmentation of smokers’ macrophages and are usually associated with characteristic dust macules/nodules. A histological finding of “variant respiratory bronchiolitis” exists in non-smokers,7 consisting of macrophages containing homogeneous eosinophilic pigment rather than the brown cytoplasmic pigment of typical respiratory bronchiolitis. The significance of this finding in a handful of cases is uncertain.
The prevalence of respiratory bronchiolitis in former smokers appears to be more variable. Serial bronchoalveolar lavage (BAL) data suggest that, on average, the percentage of macrophages in BAL fluid of ex-smokers falls to the levels of lifelong non-smokers within 3 years.10 However, there is considerable individual variation, with respiratory bronchiolitis regressing within 1 year of smoking cessation in some cases but persisting for decades in others.7
The causative link between smoking and RBILD is equally striking. In the three largest clinical series,3,5,11 all patients were current smokers apart from a single non-smoker heavily exposed to solder fumes.11 In DIP, also, most patients are smokers, including 36 of 40 patients (90%) in the largest series.12 However, DIP is sometimes present in non-smokers, especially in the setting of pneumoconiosis, drug reactions or inborn errors of metabolism.13–16 Moreover, it appears that DIP may arise without a clinically overt trigger; in a recent series, 8 of 20 patients with DIP (40%) were lifelong non-smokers but no differences in histological features were disclosed when smokers and non-smokers were compared.17
HISTOLOGICAL APPEARANCES OF RESPIRATORY BRONCHIOLITIS, RBILD AND DIP
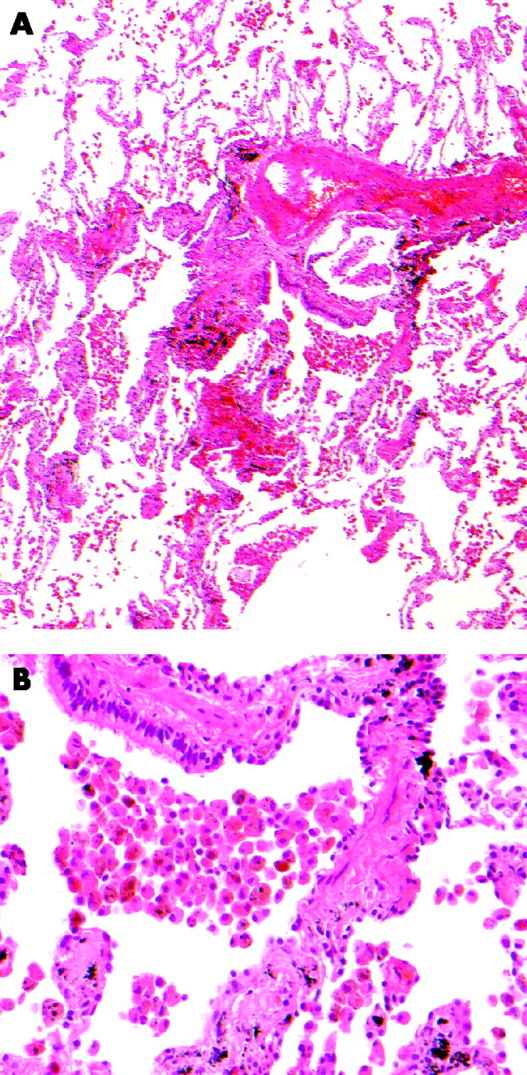
Essentially, RBILD is histologically indistinguishable from respiratory bronchiolitis,7 a view first advanced by Myers et al in the original description of RBILD.3 The cardinal feature of both disorders is the accumulation of alveolar macrophages within respiratory bronchioles, variably extending into neighbouring alveoli. Macrophages are characterised by glassy eosinophilic cytoplasm, usually with brown and finely granular pigmentation (representing constituents of cigarette smoke). There is often a chronic inflammatory cell infiltrate in bronchiolar and surrounding alveolar walls. Peribronchial alveolar septal thickening by collagen deposition radiating from the involved bronchiole is a more variable feature.7,18 Neither honeycomb change nor fibroblastic foci are features of either respiratory bronchiolitis or RBILD and, when present, should raise the possibility of a coexistent disease process. The pulmonary parenchyma distant from involved respiratory bronchioles is normal except when there are smoking-related emphysematous changes (fig 1).19
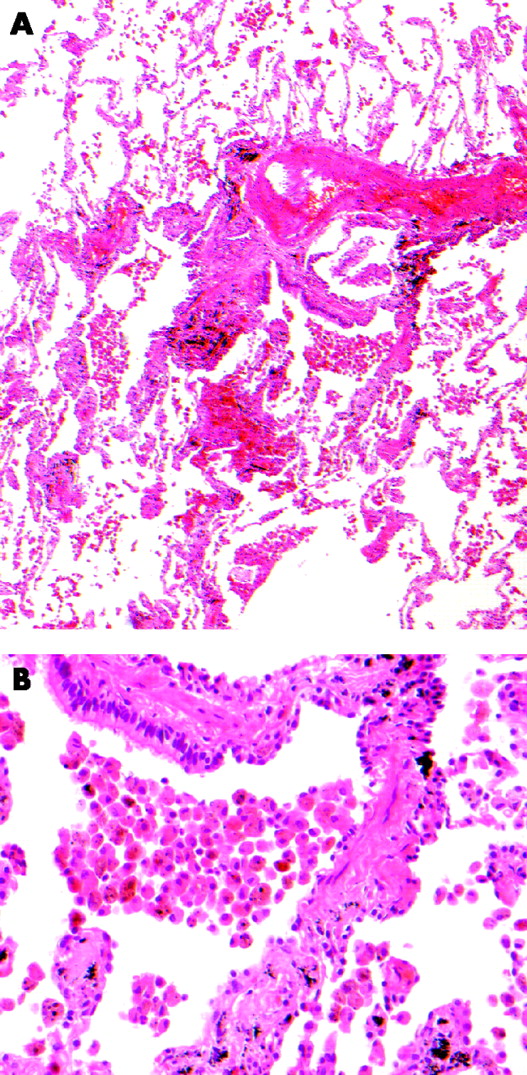
(A) Case of respiratory bronchiolitis showing accumulation of macrophages within a bronchiole and adjacent alveoli with mild peribronchiolar interstitial fibrosis. (B) Macrophages showing finely granular brown pigmentation.
Attempted histological distinctions between respiratory bronchiolitis and RBILD have centred on overall disease severity and the prominence of interstitial abnormalities, especially interstitial fibrosis.20 In several series the diagnosis of RBILD has been primarily histological.5,11,20 However, both the above criteria are fundamentally flawed. Sampling error is a particular problem. Biopsy appearances are not always a reliable guide to disease severity elsewhere in the lungs, especially if high resolution CT (HRCT) appearances are used to direct the surgeon to focal abnormalities in otherwise limited respiratory bronchiolitis. More importantly, airway centred parenchymal fibrosis is frequently present in the respiratory bronchiolitis of asymptomatic current or ex-smokers, with peribronchial fibrosis present in 9 and alveolar septal fibrosis present in 47 of 109 patients in the series of Fraig et al.7 Respiratory bronchiolitis was occasionally associated with prominent peribronchiolar fibrosis which was surprisingly silent clinically. Although population differences between the two disorders probably do exist in the average severity of disease at biopsy, these can be highly misleading in individual cases. Thus, as discussed later, distinctions between respiratory bronchiolitis and RBILD should be based on global markers of disease severity (symptoms, pulmonary function impairment, HRCT abnormalities) and not on biopsy evaluation in isolation.
Comparisons of the severity of respiratory bronchiolitis between current and former smokers have disclosed surprisingly little difference between the two subgroups.7,21 This observation may be indicative of a true dichotomy following cessation of smoking, with rapid regression of respiratory bronchiolitis in some cases but persistent disease in an important subgroup.
DIP was first described by Liebow in 1965. The histological hallmarks of DIP are a prominent accumulation of intra-alveolar macrophages, hyperplasia of type II pneumocytes and, more variably, diffuse alveolar septal thickening.22 In contrast to RBILD, macrophages are diffuse in distribution although typically containing pigment identical to that seen in RBILD and respiratory bronchiolitis in smoking-related DIP.23 Thus, the key difference between DIP and RBILD/respiratory bronchiolitis is the profusion of macrophage infiltration, prompting some to argue that these disorders are part of a common spectrum of smoking-related lung damage (fig 2).6,20

(A) Case of desquamative interstitial pneumonia showing extensive and diffuse accumulation of macrophages within alveoli. (B) Macrophages show similar pigmentation to the case of respiratory bronchiolitis shown in fig 1, although in this case there is more interstitial inflammation and greater eosinophil numbers in association with the macrophages.
IMAGING OF RESPIRATORY BRONCHIOLITIS, RBILD AND DIP
In current smokers and in ex-smokers with respiratory bronchiolitis, chest radiographic appearances are often normal and any abnormalities tend to be limited and non-specific. Abnormal features tend to be more obvious in RBILD, with bronchial wall thickening and patchy ground glass attenuation in most patients in one series24 and reticulonodular opacities in the majority of cases.3,5 However, neither RBILD nor DIP can be diagnosed with any confidence on chest radiography.
The spectrum of HRCT abnormalities associated with cigarette smoking was first defined by Remy-Jardin et al in a study of 175 asymptomatic adults, including 98 current smokers and 26 ex-smokers.25 HRCT features seen in current smokers included parenchymal micronodules (27%), areas of ground glass attenuation (21%), dependent areas of decreased attenuation (34%) and emphysema (21%), and all were significantly more prevalent than in non-smokers. Parenchymal micronodules 2–3 mm in diameter were more profuse in the upper zones and tended to be ill defined. The presence of emphysema was functionally significant but no other HRCT abnormality was associated with pulmonary function impairment. In a subsequent histological study of 41 smokers undergoing resections of solitary pulmonary nodules,9 ground glass attenuation on HRCT scanning corresponded to an accumulation of pigmented macrophages and mucus in the alveolar spaces (ie, a macrophage alveolitis), sometimes associated with mild interstitial inflammation and/or fibrosis. Poorly defined parenchymal micronodules were thought to indicate bronchiolectasis with peribronchiolar fibrosis. Thus, the HRCT abnormalities of respiratory bronchiolitis are those of inflammation and/or fibrosis in and around terminal bronchioles.26
It is now clear that the spectrum of HRCT appearances in RBILD is exactly that of respiratory bronchiolitis, although abnormalities are more extensive in RBILD. HRCT findings in early clinicopathological series of RBILD include centrilobular micronodules, ground glass attenuation, atelectasis, linear reticular abnormalities and emphysema.20,27–29 The discrepant observation in one series that most patients with a histological diagnosis of RBILD had normal HRCT appearances30 probably reflects the difficulties in distinguishing between respiratory bronchiolitis and RBILD from biopsy appearances in isolation. In the largest HRCT series of patients with a clinicopathological diagnosis of RBILD (n = 21), the most frequent HRCT features were central bronchial wall thickening proximal to segmental bronchi (n = 19, 90%), peripheral bronchial wall thickening distal to segmental bronchi (n = 18, 86%), centrilobular nodules (n = 15, 71%) and areas of ground glass opacity (n = 12, 57%).24 In earlier reports bronchial wall thickening had not been highlighted, probably because of its non-specific nature. Both centrilobular nodules and ground glass opacity tended to be diffuse with no zonal predominance, unlike smokers with HRCT evidence of respiratory bronchiolitis. Emphysema (n = 12, 57%) and patchy areas of hypoattenuation, seen predominantly in the lower lobes (n = 8, 38%), were also observed. As reported earlier in asymptomatic smokers,9 the extent of centrilobular nodules was linked to the amount of macrophage accumulation within respiratory bronchioles and the severity of chronic inflammation within respiratory bronchiolar walls. The extent of ground glass attenuation was indicative of the intensity of macrophage accumulation in the alveoli and alveolar ducts.
Areas of hypoattenuation on inspiratory HRCT scans are present in a few cases of RBILD24 and were not documented in earlier series.20,25,26 By contrast, patchy decreased attenuation is a frequent feature on expiratory HRCT scans, and this applies equally to RBILD and to asymptomatic smokers and ex-smokers.31,32 In a large series, segmental air trapping was significantly more prevalent in current smokers (26%) and ex-smokers (27%) than in non-smokers, whereas limited lobular air trapping was highly non-specific. As in earlier reports, ill defined micronodules, areas of ground glass attenuation and emphysema were frequently seen in smokers; in current smokers air trapping was often present in regions with profuse micronodular abnormalities. Patchy hypoattenuation on expiratory HRCT scans (termed “mosaic attenuation”) is a cardinal feature of small airways disease and is seen also in constrictive bronchiolitis,33 hypersensitivity pneumonitis,34 bronchiectasis35 and sarcoidosis.36
Because of the low prevalence of DIP in the modern era, the HRCT appearances of DIP have been characterised in only one moderately large series (n = 22) and this required the amalgamation of cases from several centres.37 The predominant abnormality was ground glass attentuation which was always present and usually bilateral, moderately symmetrical, peripheral and predominantly lower zone in distribution (unlike the non-regional nature of patchy ground glass attenuation in RBILD20,24). Predominantly basal irregular linear opacities, seen in over half the cases, were associated with anatomical distortion, traction bronchiectasis and, variably, with small peripheral cystic spaces thought to denote dilated bronchioles and alveolar ducts. These features, considered to represent pulmonary fibrosis, were usually limited in extent, without the honeycomb change typically present in usual interstitial pneumonia (UIP).
The reported HRCT abnormalities in respiratory bronchiolitis, RBILD and DIP can now be integrated into the clinical evaluation of these disorders. It is clear that respiratory bronchiolitis and RBILD cannot be distinguished solely from the nature of HRCT abnormalities, but that the extent of disease on HRCT scanning should be taken into account in reaching the judgement that clinically significant diffuse lung disease (ie, RBILD) is present. There are two important caveats. The mere presence of limited but compatible HRCT abnormalities does not equate with a diagnosis of RBILD: HRCT abnormalities in large series of smokers25,31,32 relate to respiratory bronchiolitis and not to RBILD. Equally importantly, there is no arbitrary cut-off in HRCT disease extent at which respiratory bronchiolitis becomes RBILD; in this regard, HRCT severity should not be evaluated in isolation but should be integrated with the degree of pulmonary function impairment and clinical severity. By contrast, HRCT features can be useful in identifying probable DIP rather than RBILD, based upon the nature, distribution and extent of ground glass attenuation as well as the presence of peripheral cystic spaces. However, the overlapping features on HRCT scans in some cases20 and the occasional histological coexistence of DIP and RBILD should be emphasised.
PULMONARY FUNCTION TESTS IN RBILD AND DIP
Despite the bronchiolocentric nature of disease in RBILD, both restrictive and obstructive abnormalities have been documented,3,5,11 with a mixed predominantly restrictive ventilatory defect—usually associated with a mild to moderate reduction in the carbon monoxide transfer factor (Tlco)—the most frequent finding. Airflow obstruction is usually mild. The severity of pulmonary function impairment is a crucial feature in distinguishing RBILD from respiratory bronchiolitis. Some patients considered to have respiratory bronchiolitis, with little or no pulmonary function impairment, have moderately extensive fibrotic change radiating from the bronchioles at lung biopsy.7 However, this is likely to reflect the problem of sampling error,38 with severity at biopsy not representative of the global severity of lung disease.
The interpretation of pulmonary function tests is more straightforward in DIP. The ventilatory defect is consistently restrictive with the reduction in Tlco a useful guide to the underlying severity of the disease. Hypoxia appears to supervene only when disease is advanced.
An important confounder in the interpretation of pulmonary function tests in both RBILD and DIP is the frequent coexistence of centrilobular emphysema. It is well recognised that emphysema is more extensive histologically than is apparent on HRCT scanning. Thus, in some patients with RBILD or DIP, a predominantly obstructive defect may reflect the presence of apparently limited emphysema. In other cases the coexistence of emphysema and fibrotic abnormalities may give rise to paradoxically normal volumes, classically seen when emphysema is associated with the more extensive pulmonary fibrosis of idiopathic pulmonary fibrosis.39 This may explain why there is often a disproportionate reduction in Tlco, which was <60% of predicted in over half the cases in two series.3,11
The severity of pulmonary function impairment is a crucial part of the distinction between respiratory bronchiolitis and RBILD. However, for the reasons discussed above, the pattern and level of functional impairment should not be used in isolation to identify RBILD without reference to clinical and HRCT findings. Furthermore, occasional patients with RBILD, as judged by clinical and HRCT criteria, have normal pulmonary function tests.18 This apparent paradox is likely to reflect the wide range of normality (80–120% of predicted values); in some patients, pulmonary function indices remain in the normal range despite considerable reductions with the development of RBILD.
CLINICAL FEATURES, PROGNOSIS AND TREATED COURSE OF RBILD AND DIP
RBILD most often presents between the third and sixth decades with insidious exertional dyspnoea and persistent cough which may be non-productive.3,5,11,19 There is no gender predilection. Chest pain and weight loss are less frequent. Occasional reports of fever and haemoptysis may represent coincidental lower respiratory tract infection. Bilateral end inspiratory crackles, which may be predominantly basal, are common but clubbing is very rare.11 The presence of symptoms is not invariable; RBILD may be diagnosed in asymptomatic patients with functional impairment and chest radiographic or HRCT abnormalities.
Compared with other forms of idiopathic interstitial pneumonia, RBILD is a benign disorder with a much better survival than UIP or fibrotic non-specific interstitial pneumonia (NSIP),40 but there are few reports of serial pulmonary function trends. In the first description of RBILD, all six patients improved or remained stable in the next 3 years.3 However, longitudinal behaviour is heterogeneous. In one series there was regression of disease in only 3 of 10 cases, with the remainder not improving functionally or, in 3 cases, having a decline in forced vital capacity or Tlco at a mean of follow-up of 4 years despite cessation of smoking and treatment.11 Disease has regressed with smoking cessation after failure of cortico-steroid and cyclophosphamide therapy.41 Partial regression of bronchial wall thickening, ground glass attenuation and centrilobular micronodules has been observed on HRCT scans following smoking cessation and treatment.24
It is not yet clear how often residual functional impairment after cessation of smoking represents residual RBILD as opposed to other smoking-related processes. The respiratory bronchiolitis invariably present in a current smoker eventually regresses in most instances with smoking cessation,7 but this observation may not apply to RBILD; although inflammation may abate with time, the fibrotic component may result in important residual functional impairment. There are anecdotal reports, including a case known to the authors, of extensive underlying fine fibrosis at biopsy not apparent from HRCT appearances. In other cases there is evidence of coexistent emphysema or fibrotic lung disease, and major improvements in pulmonary function indices may be unattainable. For all these reasons it appears likely that a significant functional improvement following smoking cessation or treatment in RBILD may be confined to a minority or, at best, a small majority of patients, although the paucity of published longitudinal data should be stressed.
Patients with DIP generally present in the fourth to sixth decade with insidious dyspnoea, non-productive cough and, less frequently, fatigue and weight loss.12 Bilateral basal inspiratory crackles are frequent whereas clubbing is rare. Like RBILD, DIP has a better outcome than UIP12 or fibrotic NSIP,40 with most patients responding to corticosteroid therapy.12,40 However, the outcome is poor in a minority of cases.12 It is likely that, in occasional cases, a poor response to corticosteroids or immunosuppressive agents is indicative of significant underlying fibrosis, especially in long standing disease. However, there are anecdotal reports of patients in whom apparently reversible macrophage predominant disease at biopsy does not regress with treatment.
KEY CLINICAL ISSUES IN RBILD AND DIP
Diagnosis
The formulation of a diagnosis of RBILD is dependent on two considerations. First, as discussed above, the disease must be sufficiently severe to present as a clinically significant diffuse lung disease as opposed to respiratory bronchiolitis. Separately, the clinician must consider other diffuse lung diseases with similar presentations. Among the non-invasive features used to diagnose RBILD, HRCT findings most often provide discriminatory information. Symptoms, clinical signs and the pattern and level of functional impairment are non-specific, except in the distinction between RBILD and respiratory bronchiolitis.
HRCT findings typical of RBILD effectively exclude most other diffuse lung diseases including the predominantly fibrotic idiopathic interstitial pneumonias (idiopathic pulmonary fibrosis (IPF), fibrotic NSIP). There is occasional overlap in appearances between RBILD and DIP, although the ground glass attenuation of DIP is usually more extensive and nodules are infrequent or absent.18,37 HRCT appearances of RBILD frequently resemble those of subacute hypersensitivity pneumonitis; in both diseases, widespread poorly formed nodular abnormalities and areas of hypoattenuation may coexist.24,34 However, the history (RBILD occurs only in smokers whereas hypersensitivity pneumonitis is rare in smokers) and BAL profile are usually definitive in distinguishing between these disorders.
In RBILD and respiratory bronchiolitis alike, a characteristic brown pigmentation of macrophages has been consistently observed. In an evaluation of patients with biopsy proven RBILD, DIP, UIP or fibrotic NSIP, patients with RBILD were distinguished by significant increases in macrophage numbers and significantly lower percentages of other cellular components.42 A BAL neutrophilia or eosinophilia is rare in RBILD. Moreover, the BAL findings in RBILD differ strikingly from those of hypersensitivity pneumonitis in which a BAL lymphocytosis is expected.43 BAL findings, in conjunction with the history and HRCT appearances, usually allow the diagnosis of RBILD to be made without the need for thoracoscopic lung biopsy.
By contrast, a thoracoscopic biopsy is almost invariably required to diagnose DIP as the HRCT appearance of extensive ground glass attenuation and the highly cellular BAL profile42 are both non-specific.
Treatment
In RBILD, in the absence of longitudinal data, management is largely driven by the severity of pulmonary function impairment. In mild to moderate disease, a period of observation after smoking cessation is usually warranted to allow spontaneous regression of disease, as almost always occurs in patients with respiratory bronchiolitis.7 A trial of corticosteroid, with or without immunosuppressive drugs, is usual when significant disease does not regress following smoking cessation.3,5,11,19,24 However, the risk:benefit ratio of prolonged treatment (with attendant side effects) merits careful consideration. Early withdrawal of treatment in non-responders is often appropriate. The same general principles apply in DIP, although early treatment is usually required as the level of functional impairment tends to be more severe than in RBILD.
ARE RBILD AND DIP THE ENDS OF A COMMON DISEASE SPECTRUM?
It is sometimes argued6,20 that RBILD and DIP should be reclassified as a single entity, based on the overlapping histological and HRCT features and a common aetiological relationship to smoking. However, RBILD and DIP continue to be regarded as separate disorders in consensus statements44,45 and this remains logical for several reasons.
First, the presenting features and clinical course of RBILD and DIP differ. DIP is usually a more aggressive process than RBILD, with no evidence that RBILD progresses to DIP.46 The nodular abnormalities of RBILD are not an HRCT feature of DIP, and the BAL profiles of the two diseases are dissimilar.42 Moreover, the indications for treatment are often marginal in RBILD whereas a vigorous approach is usually appropriate in DIP. Based on differences in disease profiles, the retention of DIP and RBILD as separate disorders is justified. On the same grounds, distinctions are currently made between other diseases historically grouped within the entity of “idiopathic pulmonary fibrosis”.44,45
Second, as highlighted in this article, the more important disease classification issue is the separation between RBILD and respiratory bronchiolitis. This separation is not straightforward in the absence of formal severity criteria for the diagnosis of RBILD, but owes much to accumulated clinical experience which would be sacrificed by an amalgamation of RBILD with DIP.
Finally, it is possible that RBILD and DIP may evolve in very different ways. The recent observation on HRCT scanning of the evolution from respiratory bronchiolitis/RBILD to emphysema in a few cases is intriguing47 and, although the overall importance of this finding has yet to be established, further exploration of the inter-relationship between RBILD and emphysema is warranted. By contrast, limited coexistent abnormalities compatible with pulmonary fibrosis are evident on HRCT scanning in DIP in over 50% of cases20,37 but appear to be less prevalent in RBILD.5,24,27 Furthermore, follow-up HRCT appearances in some patients with DIP are most reminiscent of those seen in fibrotic NSIP.17 Until the pathogenetic significance of these observations has been clarified, DIP and RBILD should continue to be regarded as separate entities.
RELATIONSHIPS BETWEEN SMOKING AND OTHER DIFFUSE LUNG DISEASES
Among other diffuse lung diseases, evidence for smoking causation is emerging for acute eosinophilic pneumonia. There are an increasing number of case reports, especially from Japan, of the early onset of acute eosinophilic pneumonia shortly after starting smoking, most often in the young;48,49 confirmation was obtained by cigarette smoking challenge tests in several cases.50 The pathogenesis is not understood, although the early presence of a neutrophilia in the lungs or blood has focused attention on interleukin-8 as a potentially important mediator.51
The relationship between smoking and other forms of diffuse lung disease is poorly understood. Sarcoidosis,52 hypersensitivity pneumonitis53 and radiation-induced pneumonitis54 are all strikingly less prevalent in smokers than in non-smokers, but a smoking protective mechanism has yet to be identified. By contrast, current or former smokers have consistently been over-represented in IPF,55–59 with smoking found to be an independent risk factor for the development of IPF in one report.60 Intriguingly, current smokers were found to have a much longer survival than former smokers in a recent large study of IPF, an effect that remained statistically significant after adjustment for functional impairment, age and the chest radiographic severity of disease.61 However, it is not yet clear whether ongoing smoking is genuinely protective against disease progression; a “healthy smoker effect” (with smoking cessation prompted by more rapidly progressive disease) has not been excluded.
Key conclusions
-
Respiratory bronchiolitis is almost invariable in current smokers, never occurs in non-smokers and can be regarded as a highly specific physiological response to smoking.
-
Respiratory bronchiolitis and respiratory bronchiolitis-associated interstitial lung disease (RBILD) are histologically indistinguishable. A surgical biopsy may be useful when other disease processes require exclusion, but should not be used in isolation to diagnose RBILD.
-
Limited high resolution CT (HRCT) abnormalities due to respiratory bronchiolitis are fairly frequent in current smokers. In RBILD, HRCT patterns are similar but more extensive.
-
The distinction between RBILD and respiratory bronchiolitis is made solely from global disease severity. RBILD is present when the combined severity of pulmonary function impairment, HRCT findings and symptoms is indicative of a clinically significant interstitial lung disease.
-
Treatment (initially with corticosteroid therapy) is generally warranted in desquamative interstitial pneumonia (DIP) but is seldom necessary in RBILD, especially if smoking cessation is achieved.
The inter-relationship between smoking-related diffuse interstitial lung diseases is currently a subject of considerable anecdotal speculation.62 Areas of DIP-like reaction have been observed in a few asymptomatic smokers with respiratory bronchiolitis, with appearances histologically indistinguishable from DIP.7 In Langerhans’ cell histiocytosis, RIB and DIP-like changes were recently found to be invariably present in all biopsy specimens (fig 3) and were occasionally sufficiently severe to cause ground glass attenuation on HRCT scanning.63 In occasional current and former smokers, histological features of the complete range of smoking-related airway and diffuse lung diseases are seen, including Langerhans’ cell histiocytosis, RBILD, emphysema and changes of pulmonary fibrosis.18 The prevalence and implications of this phenomenon and the inter-relationships between these various disorders are only now emerging. The possible significance of NSIP in smokers with coexistent RBILD or DIP is currently being explored in a combined American Thoracic Society/European Respiratory Society workshop.

{kind=link}
{kind=link}
{kind=link}
(A) The predominant histological feature in this smoker is that of desquamative interstitial pneumonia but (B) occasional centriacinar stellate scars typical of burnt out Langerhans’ granulomatosis are present within the parenchyma.
REFERENCES
Footnotes
-
Competing interests: None.